Basic usage¶
Note
To follow this tutorial, it is assumed that MAICoS has been installed on your computer.
In this tutorial we will use the maicos.DensityPlanar to extract the density profile
of water molecules confined in a 2D slit pore from a molecular dynamics simulation.
In general, all modules follow the same structure:
load your simulation trajectory and define atom selections
define analysis parameters like bin width or the direction of the analysis
after the analysis was succesful, access all results in a
MDAnalysis.analysis.base.Resultsof the analysis object.
Important
Some of the calculations may contain pitfalls, such as dielectric profiles calculation. Potential pitfalls and best practices are listed in the How-to guides section.
MAICoS can be used equally from the Python interpreter or the CLI. The Python interpreter allows usually more versatile analysis scrcipts. But, using CLI instead of the Python interpreter can sometimes be more comfortable, particularly for lengthy standard analysis.
The documentation, almost exclusively describes the use of MAICoS from the Python interpreter, but all operations can be equivalently performed from the CLI.
Loading library¶
To start, let us first import Matplotlib, MDAnalysis and MAICoS
import matplotlib.pyplot as plt
import MDAnalysis as mda
import maicos
After Installation, any MAICoS module can be accessed simply by using the module name,
for example in this tutorial we will use maicos.DensityPlanar:
maicos densityplanar
Opening trajectory¶
For this tutorial we use a system consisting of a 2D slab with 1176 water molecules confined in a 2D slit made of NaCl atoms, where the two water/solid interfaces are normal to the axis \(z\) as shown in the snapshot below:
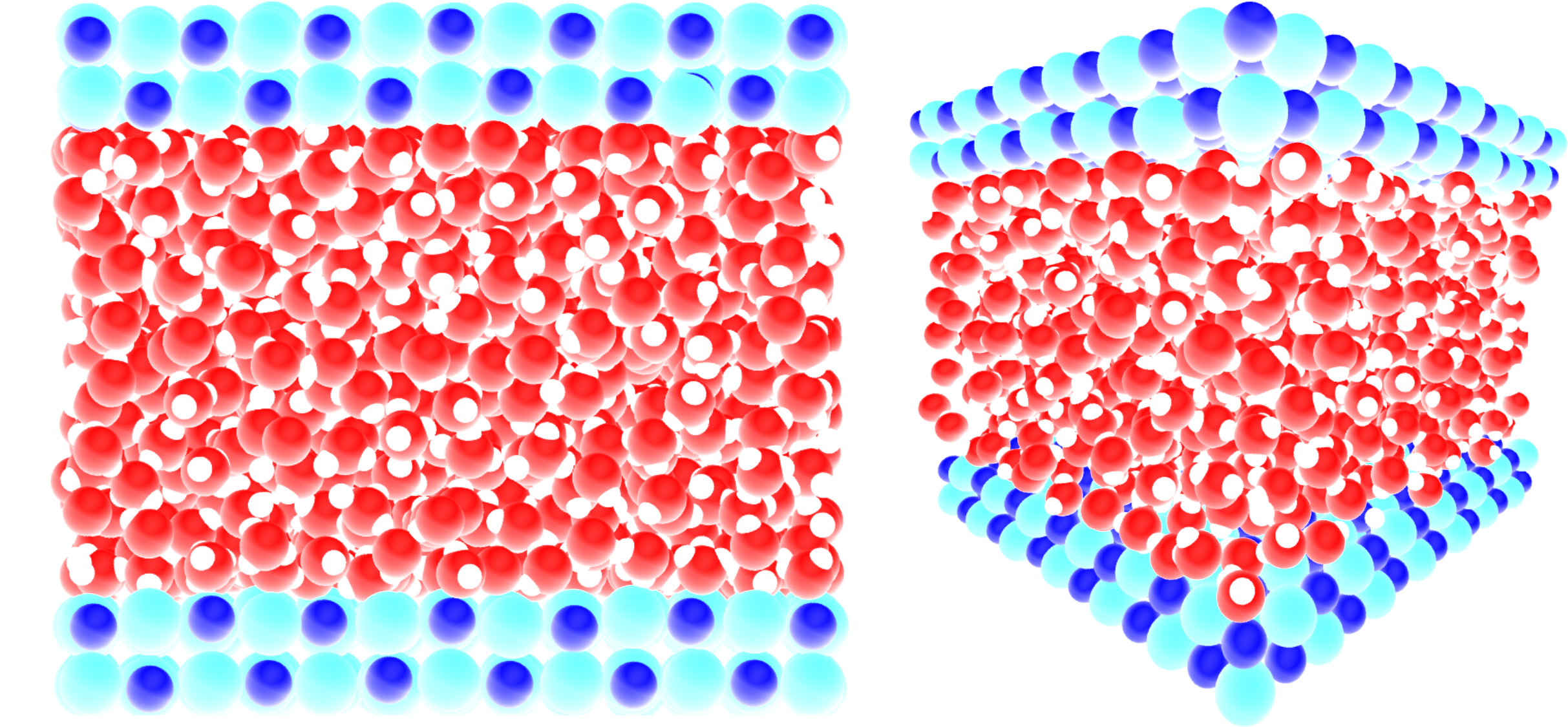

An acceleration \(a = 0.05\,\text{nm}\,\text{ps}^{-2}\) was applied to the water molecules in the \(\boldsymbol{e}_x\) direction parallel to the NaCl wall, and the atoms of the wall were maintained frozen along \(\boldsymbol{e}_x\).
You can download the required topology and the trajectory from our website.
We first create an MDAnalysis.core.universe.Universe by loading a topology
and trajectory from disk.
u = mda.Universe("slit_flow.tpr", "slit_flow.trr")
Let us print some information about the trajectory:
print(f"Number of frames in the trajectory is {u.trajectory.n_frames}.")
timestep = round(u.trajectory.dt, 2)
print(f"Time interval between two frames is {timestep} ps.")
total_time = round(u.trajectory.totaltime, 2)
print(f"Total simulation time is {total_time} ps.")
Number of frames in the trajectory is 201.
Time interval between two frames is 10.0 ps.
Total simulation time is 2000.0 ps.
We can specify the topology and trajectory using the -s and -f flags.
maicos densityplanar -s slit_flow.tpr -f slit_flow.trr
Selecting Subsets of Atoms¶
Now, we define an atom group containing the oxygen and the hydrogen atoms (of the water molecules).
Now, we define an atomgroup containing the oxygen and the hydrogen atoms:
group_H2O = u.select_atoms("type OW HW")
Let us print some information about the groups
print(f"Number of water molecules is {group_H2O.n_atoms // 3}.")
Number of water molecules is 1176.
Using the -atomgroup flag.
maicos densityplanar -s slit_flow.tpr \
-f slit_flow.trr \
-atomgroup "type OW HW"
The density profile has been written in a file named density.dat in the current
directory. The written file starts with the following lines
head -n 20 density.dat
Running an Analysis¶
Let us use now finally use the maicos.DensityPlanar class to extract the
density profile along the (default) \(z\) axis by running the analysis.
dplan = maicos.DensityPlanar(group_H2O).run()
The warning starting with Unwrapping is expected and can be ignored for now.
Let us extract the bin coordinates \(z\), the averaged density profile and its
uncertainty estimated by MAICoS from the results attribute:
zcoor = dplan.results.bin_pos
dens = dplan.results.profile
uncertainity = dplan.results.dprofile
The density profile is given as a 1D array, let us look at the 10 first lines:
print(dens[:10])
[0.00000000e+00 0.00000000e+00 0.00000000e+00 0.00000000e+00
0.00000000e+00 2.91778321e-05 3.26228275e-01 9.99460390e-01
5.32837124e-01 5.64829866e-01]
By default the bin_width is 1 Å, and the unit is atomic mass per \(Å^3\)
(\(\text{u}/\text{Å}^3\)).
For lengthy analysis, use the concfreq option to update the result during the run
maicos densityplanar -s slit_flow.tpr \
-f slit_flow.trr \
-atomgroup 'type OW HW' \
-concfreq '10'
Visualizing Results¶
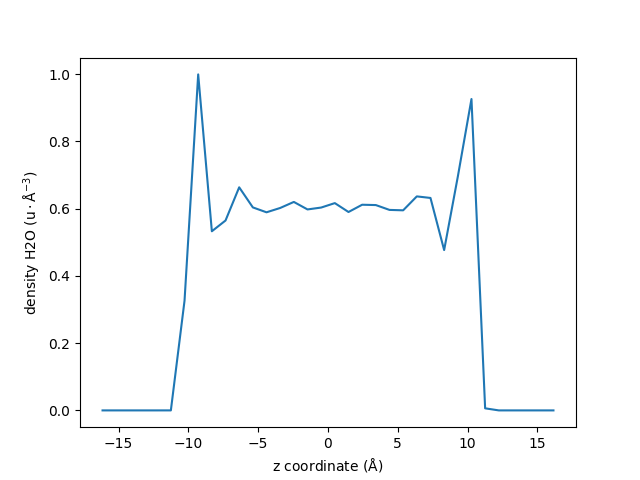
We can now plot the density profile.
Using Matplotlib:
fig, ax = plt.subplots()
ax.errorbar(zcoor, dens, 5 * uncertainity)
ax.set_xlabel(r"z coordinate ($\rm Å$)")
ax.set_ylabel(r"density H2O ($\rm u \cdot Å^{-3}$)")
fig.show()
Using gnuplot:
echo " \
set xlabel 'z coordinate, (Å)'; \
set ylabel 'density H2O (u.Å⁻³)'; \
plot 'density.dat' using (column(1)):(column(2)):(5*column(3)) with yerrorlines title '' \
" | gnuplot || true
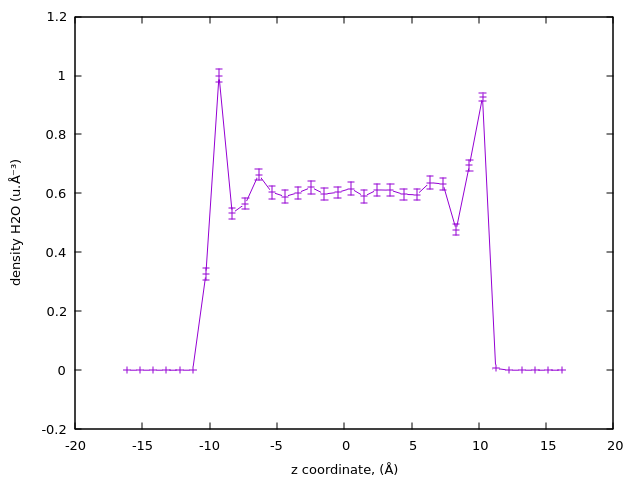
For this example we scales the error by 5 to be visible in the plot. More details on the uncertainty estimation can be found in Advanced usage.
Getting Help¶
MAICoS provides help both for the main program and for each analysis module.
The general help of MAICoS can be accessed using
help(maicos)
Help on package maicos:
NAME
maicos - MAICoS: Molecular Analysis of Interfacial and COnfined Systems.
PACKAGE CONTENTS
__main__
_version
core (package)
lib (package)
modules (package)
CLASSES
builtins.object
maicos.modules.dielectricspectrum.DielectricSpectrum
maicos.modules.dipoleangle.DipoleAngle
maicos.modules.diporderstructurefactor.DiporderStructureFactor
maicos.modules.kineticenergy.KineticEnergy
maicos.modules.rdfdiporder.RDFDiporder
maicos.modules.saxs.Saxs
maicos.core.cylinder.CylinderBase(maicos.core.planar.PlanarBase)
maicos.modules.dielectriccylinder.DielectricCylinder
maicos.modules.pdfcylinder.PDFCylinder
maicos.core.cylinder.ProfileCylinderBase(maicos.core.cylinder.CylinderBase, maicos.core.base.ProfileBase)
maicos.modules.densitycylinder.DensityCylinder
maicos.modules.dipordercylinder.DiporderCylinder
maicos.modules.velocitycylinder.VelocityCylinder
maicos.core.planar.PlanarBase(maicos.core.base.AnalysisBase)
maicos.modules.dielectricplanar.DielectricPlanar
maicos.modules.pdfplanar.PDFPlanar
maicos.core.planar.ProfilePlanarBase(maicos.core.planar.PlanarBase, maicos.core.base.ProfileBase)
maicos.modules.densityplanar.DensityPlanar
maicos.modules.diporderplanar.DiporderPlanar
maicos.modules.temperatureplanar.TemperaturePlanar
maicos.modules.velocityplanar.VelocityPlanar
maicos.core.sphere.ProfileSphereBase(maicos.core.sphere.SphereBase, maicos.core.base.ProfileBase)
maicos.modules.densitysphere.DensitySphere
maicos.modules.dipordersphere.DiporderSphere
maicos.core.sphere.SphereBase(maicos.core.base.AnalysisBase)
maicos.modules.dielectricsphere.DielectricSphere
class DensityCylinder(maicos.core.cylinder.ProfileCylinderBase)
| DensityCylinder(
| atomgroup: mda.AtomGroup,
| dens: str = 'mass',
| dim: int = 2,
| zmin: float | None = None,
| zmax: float | None = None,
| rmin: float = 0,
| rmax: float | None = None,
| bin_width: float = 1,
| bin_method: str = 'com',
| grouping: str = 'atoms',
| refgroup: mda.AtomGroup | None = None,
| unwrap: bool = True,
| pack: bool = True,
| concfreq: int = 0,
| jitter: float = 0.0,
| output: str = 'density.dat'
| ) -> None
|
| Cylindrical partial density profiles.
|
| Calculations are carried out for ``mass``
| :math:`(\rm u \cdot Å^{-3})`, ``number`` :math:`(\rm Å^{-3})`, partial ``charge``
| :math:`(\rm e \cdot Å^{-3})` or electron :math:`(\rm e \cdot Å^{-3})` density
| profiles
| along the radial axis in a cylindrical coordinate system,
| with principal axis along ``[x, y, z]`` axes of the simulation cell. The origin of the
| coordinate system defaults to the box center, but can be set to dynamically follow the
| center of mass of a reference group via ``refgroup``
| Cell dimensions are allowed to fluctuate in time.
|
| For grouping with respect to ``molecules``, ``residues`` etc., the corresponding
| centers (i.e., center of mass), taking into account periodic boundary conditions,
| are calculated. For these calculations molecules will be unwrapped/made whole.
| Trajectories containing already whole molecules can be run with ``unwrap=False`` to
| gain a speedup. For grouping with respect to atoms, the ``unwrap`` option is always
| ignored.
|
| For the correlation analysis the 0th bin of the 0th's
| group profile is used. For further information on the correlation analysis please
| refer to :class:`AnalysisBase <maicos.core.base.AnalysisBase>` or the
| :ref:`general-design` section.
|
| Parameters
| ----------
| atomgroup : MDAnalysis.core.groups.AtomGroup
| A :class:`~MDAnalysis.core.groups.AtomGroup` for which the calculations are
| performed.
| dens : {``"mass"``, ``"number"``, ``"charge"``, ``"electron"``}
| density type to be calculated.
| dim : {0, 1, 2}
| Dimension for binning (``x=0``, ``y=1``, ``z=1``).
| zmin : float
| Minimal coordinate for evaluation (in Å) with respect to the center of mass of
| the refgroup.
| If ``zmin=None``, all coordinates down to the lower cell boundary are taken into
| account.
| zmax : float
| Maximal coordinate for evaluation (in Å) with respect to the center of mass of the
| refgroup.
| If ``zmax = None``, all coordinates up to the upper cell boundary are taken into
| account.
| rmin : float
| Minimal radial coordinate relative to the center of mass of the refgroup for
| evaluation (in Å).
| rmax : float
| Maximal radial coordinate relative to the center of mass of the refgroup for
| evaluation (in Å).
|
| If ``rmax=None``, the box extension is taken.
| bin_width : float
| Width of the bins (in Å).
| bin_method : {``"com"``, ``"cog"``, ``"coc"``}
| Method for the position binning.
|
| The possible options are center of mass (``"com"``), center of geometry (``"cog"``),
| and center of charge (``"coc"``).
| grouping : {``"atoms"``, ``"residues"``, ``"segments"``, ``"molecules"``, ``"fragments"``}
| Atom grouping for the calculations.
|
| The possible grouping options are the atom positions (in the case where
| ``grouping="atoms"``) or the center of mass of the specified grouping unit (in the
| case where ``grouping="residues"``, ``"segments"``, ``"molecules"`` or
| ``"fragments"``).
| refgroup : MDAnalysis.core.groups.AtomGroup
| Reference :class:`~MDAnalysis.core.groups.AtomGroup` used for the calculation. If
| ``refgroup`` is provided, the calculation is performed relative to the center of
| mass of the AtomGroup. If ``refgroup`` is :obj:`None` the calculations are performed
| with respect to the center of the (changing) box.
| unwrap : bool
| When :obj:`True`, molecules that are broken due to the periodic boundary conditions
| are made whole.
|
| If the input contains molecules that are already whole, speed up the calculation by
| disabling unwrap. To do so, use the flag ``-no-unwrap`` when using MAICoS from the
| command line, or use ``unwrap=False`` when using MAICoS from the Python interpreter.
|
| Note: Molecules containing virtual sites (e.g. TIP4P water models) are not currently
| supported in MDAnalysis. In this case, you need to provide unwrapped trajectory
| files directly, and disable unwrap. Trajectories can be unwrapped, for example,
| using the ``trjconv`` command of GROMACS.
| pack : bool
| When :obj:`True`, molecules are put back into the unit cell. This is required
| because MAICoS only takes into account molecules that are inside the unit cell.
|
| If the input contains molecules that are already packed, speed up the calculation by
| disabling packing with ``pack=False``.
| jitter : float
| Magnitude of the random noise to add to the atomic positions.
|
| A jitter can be used to stabilize the aliasing effects sometimes appearing when
| histogramming data. The jitter value should be about the precision of the
| trajectory. In that case, using jitter will not alter the results of the histogram.
| If ``jitter = 0.0`` (default), the original atomic positions are kept unchanged.
|
| You can estimate the precision of the positions in your trajectory with
| :func:`maicos.lib.util.trajectory_precision`. Note that if the precision is not the
| same for all frames, the smallest precision should be used.
| concfreq : int
| When concfreq (for conclude frequency) is larger than ``0``, the conclude function
| is called and the output files are written every ``concfreq`` frames.
| output : str
| Output filename.
|
| Attributes
| ----------
| results.bin_pos : numpy.ndarray
| Bin positions (in Å) ranging from ``rmin`` to ``rmax``.
| results.profile : numpy.ndarray
| Calculated profile.
| results.dprofile : numpy.ndarray
| Estimated profile's uncertainity.
|
| Method resolution order:
| DensityCylinder
| maicos.core.cylinder.ProfileCylinderBase
| maicos.core.cylinder.CylinderBase
| maicos.core.planar.PlanarBase
| maicos.core.base.AnalysisBase
| maicos.core.base._Runner
| MDAnalysis.analysis.base.AnalysisBase
| maicos.core.base.ProfileBase
| builtins.object
|
| Methods defined here:
|
| __init__(
| self,
| atomgroup: mda.AtomGroup,
| dens: str = 'mass',
| dim: int = 2,
| zmin: float | None = None,
| zmax: float | None = None,
| rmin: float = 0,
| rmax: float | None = None,
| bin_width: float = 1,
| bin_method: str = 'com',
| grouping: str = 'atoms',
| refgroup: mda.AtomGroup | None = None,
| unwrap: bool = True,
| pack: bool = True,
| concfreq: int = 0,
| jitter: float = 0.0,
| output: str = 'density.dat'
| ) -> None
| Initialize self. See help(type(self)) for accurate signature.
|
| ----------------------------------------------------------------------
| Readonly properties inherited from maicos.core.planar.PlanarBase:
|
| odims
| Other dimensions perpendicular to dim i.e. (0,2) if dim = 1.
|
| ----------------------------------------------------------------------
| Methods inherited from maicos.core.base.AnalysisBase:
|
| dump(self, filename: str) -> None
| Save analysis state to an ``.npz`` file.
|
| .. warning::
|
| :meth:`dump` is **not** an archival format. The on-disk layout
| tracks the installed MAICoS and MDAnalysis versions and may stop
| loading after an upgrade. Use it to checkpoint or hand off an
| in-progress analysis, not to preserve results long-term. For
| archival output write summary tables with :meth:`save` or export
| :attr:`results` to a stable format.
|
| Persists all statistical accumulators (``means``, ``sems``, ``sums``,
| ``pop``, ``M2``), the ``results`` and ``_obs`` containers, per-frame
| arrays (``timeseries``, ``frames``, ``times``), metadata, the associated
| Universe and the analysed atomgroup. Restore the analysis with :meth:`load`.
|
| Parameters
| ----------
| filename : str
| Path to the output ``.npz`` file.
|
| run(
| self,
| start: int | None = None,
| stop: int | None = None,
| step: int | None = None,
| frames: int | None = None,
| verbose: bool | None = None,
| progressbar_kwargs: dict | None = None
| ) -> Self
| Iterate over the trajectory.
|
| Parameters
| ----------
| start : int
| start frame of analysis
| stop : int
| stop frame of analysis
| step : int
| number of frames to skip between each analysed frame
| frames : array_like
| array of integers or booleans to slice trajectory; ``frames`` can only be
| used *instead* of ``start``, ``stop``, and ``step``. Setting *both*
| ``frames`` and at least one of ``start``, ``stop``, ``step`` to a
| non-default value will raise a :exc:`ValueError`.
| verbose : bool
| Turn on verbosity
| progressbar_kwargs : dict
| ProgressBar keywords with custom parameters regarding progress bar position,
| etc; see :class:`MDAnalysis.lib.log.ProgressBar` for full list.
|
| Returns
| -------
| self : object
| analysis object
|
| savetxt(self, fname: str, X: np.ndarray, columns: list[str] | None = None) -> None
| Save to text.
|
| An extension of the numpy savetxt function. Adds the command line input to the
| header and checks for a doubled defined filesuffix.
|
| Return a header for the text file to save the data to. This method builds a
| generic header that can be used by any MAICoS module. It is called by the save
| method of each module.
|
| The information it collects is:
| - timestamp of the analysis
| - name of the module
| - version of MAICoS that was used
| - command line arguments that were used to run the module
| - module call including the default arguments
| - number of frames that were analyzed
| - atomgroup that was analyzed
| - output messages from modules and base classes (if they exist)
|
| ----------------------------------------------------------------------
| Class methods inherited from maicos.core.base.AnalysisBase:
|
| load(filename: str) -> Self
| Restore an analysis instance from a file created by :meth:`dump`.
|
| Returns a new instance of ``cls`` with all statistical accumulators,
| per-frame arrays, metadata, and the analysed atomgroup populated from
| the file. The rebuilt :class:`MDAnalysis.Universe` carries only the
| topology — no trajectory — so calling :meth:`run` on the returned
| instance raises :class:`RuntimeError`. Use the loaded instance to
| inspect :attr:`results` or to call :meth:`save`.
|
| Parameters
| ----------
| filename : str
| Path to the ``.npz`` file written by :meth:`dump`.
|
| Returns
| -------
| Self
| A new instance of ``cls`` with state restored from ``filename``.
|
| ----------------------------------------------------------------------
| Readonly properties inherited from maicos.core.base.AnalysisBase:
|
| box_center
| Center of the simulation cell.
|
| box_lengths
| Lengths of the simulation cell vectors.
|
| ----------------------------------------------------------------------
| Data descriptors inherited from maicos.core.base._Runner:
|
| __dict__
| dictionary for instance variables
|
| __weakref__
| list of weak references to the object
|
| ----------------------------------------------------------------------
| Class methods inherited from MDAnalysis.analysis.base.AnalysisBase:
|
| get_supported_backends()
| Tuple with backends supported by the core library for a given class.
| User can pass either one of these values as ``backend=...`` to
| :meth:`run()` method, or a custom object that has ``apply`` method
| (see documentation for :meth:`run()`):
|
| - 'serial': no parallelization
| - 'multiprocessing': parallelization using `multiprocessing.Pool`
| - 'dask': parallelization using `dask.delayed.compute()`. Requires
| installation of `mdanalysis[dask]`
|
| If you want to add your own backend to an existing class, pass a
| :class:`backends.BackendBase` subclass (see its documentation to learn
| how to implement it properly), and specify ``unsupported_backend=True``.
|
| Returns
| -------
| tuple
| names of built-in backends that can be used in :meth:`run(backend=...)`
|
|
| .. versionadded:: 2.8.0
|
| ----------------------------------------------------------------------
| Readonly properties inherited from MDAnalysis.analysis.base.AnalysisBase:
|
| parallelizable
| Boolean mark showing that a given class can be parallelizable with
| split-apply-combine procedure. Namely, if we can safely distribute
| :meth:`_single_frame` to multiple workers and then combine them with a
| proper :meth:`_conclude` call. If set to ``False``, no backends except
| for ``serial`` are supported.
|
| .. note:: If you want to check parallelizability of the whole class, without
| explicitly creating an instance of the class, see
| :attr:`_analysis_algorithm_is_parallelizable`. Note that you
| setting it to other value will break things if the algorithm
| behind the analysis is not trivially parallelizable.
|
|
| Returns
| -------
| bool
| if a given ``AnalysisBase`` subclass instance
| is parallelizable with split-apply-combine, or not
|
|
| .. versionadded:: 2.8.0
|
| ----------------------------------------------------------------------
| Methods inherited from maicos.core.base.ProfileBase:
|
| save(self) -> None
| Save results of analysis to file specified by ``output``.
class DensityPlanar(maicos.core.planar.ProfilePlanarBase)
| DensityPlanar(
| atomgroup: mda.AtomGroup,
| dens: str = 'mass',
| dim: int = 2,
| zmin: float | None = None,
| zmax: float | None = None,
| bin_width: float = 1,
| bin_method: str = 'com',
| grouping: str = 'atoms',
| sym: bool = False,
| refgroup: mda.AtomGroup | None = None,
| unwrap: bool = True,
| pack: bool = True,
| jitter: float = 0.0,
| concfreq: int = 0,
| output: str = 'density.dat'
| ) -> None
|
| Cartesian partial density profiles.
|
| Calculations are carried out for ``mass``
| :math:`(\rm u \cdot Å^{-3})`, ``number`` :math:`(\rm Å^{-3})`, partial ``charge``
| :math:`(\rm e \cdot Å^{-3})` or electron :math:`(\rm e \cdot Å^{-3})` density
| profiles
| along certain cartesian axes ``[x, y, z]`` of the simulation
| cell.
| Cell dimensions are allowed to fluctuate in time.
|
| For grouping with respect to ``molecules``, ``residues`` etc., the corresponding
| centers (i.e., center of mass), taking into account periodic boundary conditions,
| are calculated. For these calculations molecules will be unwrapped/made whole.
| Trajectories containing already whole molecules can be run with ``unwrap=False`` to
| gain a speedup. For grouping with respect to atoms, the ``unwrap`` option is always
| ignored.
|
| For the correlation analysis the central bin
| (:math:`N / 2`) of the 0th's group profile is used. For further information on the correlation analysis please
| refer to :class:`AnalysisBase <maicos.core.base.AnalysisBase>` or the
| :ref:`general-design` section.
|
| Parameters
| ----------
| atomgroup : MDAnalysis.core.groups.AtomGroup
| A :class:`~MDAnalysis.core.groups.AtomGroup` for which the calculations are
| performed.
| dens : {``"mass"``, ``"number"``, ``"charge"``, ``"electron"``}
| density type to be calculated.
|
| dim : {0, 1, 2}
| Dimension for binning (``x=0``, ``y=1``, ``z=1``).
| zmin : float
| Minimal coordinate for evaluation (in Å) with respect to the center of mass of
| the refgroup.
| If ``zmin=None``, all coordinates down to the lower cell boundary are taken into
| account.
| zmax : float
| Maximal coordinate for evaluation (in Å) with respect to the center of mass of the
| refgroup.
| If ``zmax = None``, all coordinates up to the upper cell boundary are taken into
| account.
| bin_width : float
| Width of the bins (in Å).
| bin_method : {``"com"``, ``"cog"``, ``"coc"``}
| Method for the position binning.
|
| The possible options are center of mass (``"com"``), center of geometry (``"cog"``),
| and center of charge (``"coc"``).
| grouping : {``"atoms"``, ``"residues"``, ``"segments"``, ``"molecules"``, ``"fragments"``}
| Atom grouping for the calculations.
|
| The possible grouping options are the atom positions (in the case where
| ``grouping="atoms"``) or the center of mass of the specified grouping unit (in the
| case where ``grouping="residues"``, ``"segments"``, ``"molecules"`` or
| ``"fragments"``).
| sym : bool
| Symmetrize the profile. Only works in combination with ``refgroup``.
| refgroup : MDAnalysis.core.groups.AtomGroup
| Reference :class:`~MDAnalysis.core.groups.AtomGroup` used for the calculation. If
| ``refgroup`` is provided, the calculation is performed relative to the center of
| mass of the AtomGroup. If ``refgroup`` is :obj:`None` the calculations are performed
| with respect to the center of the (changing) box.
| unwrap : bool
| When :obj:`True`, molecules that are broken due to the periodic boundary conditions
| are made whole.
|
| If the input contains molecules that are already whole, speed up the calculation by
| disabling unwrap. To do so, use the flag ``-no-unwrap`` when using MAICoS from the
| command line, or use ``unwrap=False`` when using MAICoS from the Python interpreter.
|
| Note: Molecules containing virtual sites (e.g. TIP4P water models) are not currently
| supported in MDAnalysis. In this case, you need to provide unwrapped trajectory
| files directly, and disable unwrap. Trajectories can be unwrapped, for example,
| using the ``trjconv`` command of GROMACS.
| pack : bool
| When :obj:`True`, molecules are put back into the unit cell. This is required
| because MAICoS only takes into account molecules that are inside the unit cell.
|
| If the input contains molecules that are already packed, speed up the calculation by
| disabling packing with ``pack=False``.
| jitter : float
| Magnitude of the random noise to add to the atomic positions.
|
| A jitter can be used to stabilize the aliasing effects sometimes appearing when
| histogramming data. The jitter value should be about the precision of the
| trajectory. In that case, using jitter will not alter the results of the histogram.
| If ``jitter = 0.0`` (default), the original atomic positions are kept unchanged.
|
| You can estimate the precision of the positions in your trajectory with
| :func:`maicos.lib.util.trajectory_precision`. Note that if the precision is not the
| same for all frames, the smallest precision should be used.
| concfreq : int
| When concfreq (for conclude frequency) is larger than ``0``, the conclude function
| is called and the output files are written every ``concfreq`` frames.
| output : str
| Output filename.
|
| Attributes
| ----------
| results.bin_pos : numpy.ndarray
| Bin positions (in Å) ranging from ``zmin`` to ``zmax``.
| results.profile : numpy.ndarray
| Calculated profile.
| results.dprofile : numpy.ndarray
| Estimated profile's uncertainity.
|
| Notes
| -----
| Partial mass density profiles can be used to calculate the ideal component of the
| chemical potential. For details, take a look at the corresponding :ref:`How-to
| guide<sphx_glr_generated_examples_basics_chemical-potential.py>`.
|
| Method resolution order:
| DensityPlanar
| maicos.core.planar.ProfilePlanarBase
| maicos.core.planar.PlanarBase
| maicos.core.base.AnalysisBase
| maicos.core.base._Runner
| MDAnalysis.analysis.base.AnalysisBase
| maicos.core.base.ProfileBase
| builtins.object
|
| Methods defined here:
|
| __init__(
| self,
| atomgroup: mda.AtomGroup,
| dens: str = 'mass',
| dim: int = 2,
| zmin: float | None = None,
| zmax: float | None = None,
| bin_width: float = 1,
| bin_method: str = 'com',
| grouping: str = 'atoms',
| sym: bool = False,
| refgroup: mda.AtomGroup | None = None,
| unwrap: bool = True,
| pack: bool = True,
| jitter: float = 0.0,
| concfreq: int = 0,
| output: str = 'density.dat'
| ) -> None
| Initialize self. See help(type(self)) for accurate signature.
|
| ----------------------------------------------------------------------
| Readonly properties inherited from maicos.core.planar.PlanarBase:
|
| odims
| Other dimensions perpendicular to dim i.e. (0,2) if dim = 1.
|
| ----------------------------------------------------------------------
| Methods inherited from maicos.core.base.AnalysisBase:
|
| dump(self, filename: str) -> None
| Save analysis state to an ``.npz`` file.
|
| .. warning::
|
| :meth:`dump` is **not** an archival format. The on-disk layout
| tracks the installed MAICoS and MDAnalysis versions and may stop
| loading after an upgrade. Use it to checkpoint or hand off an
| in-progress analysis, not to preserve results long-term. For
| archival output write summary tables with :meth:`save` or export
| :attr:`results` to a stable format.
|
| Persists all statistical accumulators (``means``, ``sems``, ``sums``,
| ``pop``, ``M2``), the ``results`` and ``_obs`` containers, per-frame
| arrays (``timeseries``, ``frames``, ``times``), metadata, the associated
| Universe and the analysed atomgroup. Restore the analysis with :meth:`load`.
|
| Parameters
| ----------
| filename : str
| Path to the output ``.npz`` file.
|
| run(
| self,
| start: int | None = None,
| stop: int | None = None,
| step: int | None = None,
| frames: int | None = None,
| verbose: bool | None = None,
| progressbar_kwargs: dict | None = None
| ) -> Self
| Iterate over the trajectory.
|
| Parameters
| ----------
| start : int
| start frame of analysis
| stop : int
| stop frame of analysis
| step : int
| number of frames to skip between each analysed frame
| frames : array_like
| array of integers or booleans to slice trajectory; ``frames`` can only be
| used *instead* of ``start``, ``stop``, and ``step``. Setting *both*
| ``frames`` and at least one of ``start``, ``stop``, ``step`` to a
| non-default value will raise a :exc:`ValueError`.
| verbose : bool
| Turn on verbosity
| progressbar_kwargs : dict
| ProgressBar keywords with custom parameters regarding progress bar position,
| etc; see :class:`MDAnalysis.lib.log.ProgressBar` for full list.
|
| Returns
| -------
| self : object
| analysis object
|
| savetxt(self, fname: str, X: np.ndarray, columns: list[str] | None = None) -> None
| Save to text.
|
| An extension of the numpy savetxt function. Adds the command line input to the
| header and checks for a doubled defined filesuffix.
|
| Return a header for the text file to save the data to. This method builds a
| generic header that can be used by any MAICoS module. It is called by the save
| method of each module.
|
| The information it collects is:
| - timestamp of the analysis
| - name of the module
| - version of MAICoS that was used
| - command line arguments that were used to run the module
| - module call including the default arguments
| - number of frames that were analyzed
| - atomgroup that was analyzed
| - output messages from modules and base classes (if they exist)
|
| ----------------------------------------------------------------------
| Class methods inherited from maicos.core.base.AnalysisBase:
|
| load(filename: str) -> Self
| Restore an analysis instance from a file created by :meth:`dump`.
|
| Returns a new instance of ``cls`` with all statistical accumulators,
| per-frame arrays, metadata, and the analysed atomgroup populated from
| the file. The rebuilt :class:`MDAnalysis.Universe` carries only the
| topology — no trajectory — so calling :meth:`run` on the returned
| instance raises :class:`RuntimeError`. Use the loaded instance to
| inspect :attr:`results` or to call :meth:`save`.
|
| Parameters
| ----------
| filename : str
| Path to the ``.npz`` file written by :meth:`dump`.
|
| Returns
| -------
| Self
| A new instance of ``cls`` with state restored from ``filename``.
|
| ----------------------------------------------------------------------
| Readonly properties inherited from maicos.core.base.AnalysisBase:
|
| box_center
| Center of the simulation cell.
|
| box_lengths
| Lengths of the simulation cell vectors.
|
| ----------------------------------------------------------------------
| Data descriptors inherited from maicos.core.base._Runner:
|
| __dict__
| dictionary for instance variables
|
| __weakref__
| list of weak references to the object
|
| ----------------------------------------------------------------------
| Class methods inherited from MDAnalysis.analysis.base.AnalysisBase:
|
| get_supported_backends()
| Tuple with backends supported by the core library for a given class.
| User can pass either one of these values as ``backend=...`` to
| :meth:`run()` method, or a custom object that has ``apply`` method
| (see documentation for :meth:`run()`):
|
| - 'serial': no parallelization
| - 'multiprocessing': parallelization using `multiprocessing.Pool`
| - 'dask': parallelization using `dask.delayed.compute()`. Requires
| installation of `mdanalysis[dask]`
|
| If you want to add your own backend to an existing class, pass a
| :class:`backends.BackendBase` subclass (see its documentation to learn
| how to implement it properly), and specify ``unsupported_backend=True``.
|
| Returns
| -------
| tuple
| names of built-in backends that can be used in :meth:`run(backend=...)`
|
|
| .. versionadded:: 2.8.0
|
| ----------------------------------------------------------------------
| Readonly properties inherited from MDAnalysis.analysis.base.AnalysisBase:
|
| parallelizable
| Boolean mark showing that a given class can be parallelizable with
| split-apply-combine procedure. Namely, if we can safely distribute
| :meth:`_single_frame` to multiple workers and then combine them with a
| proper :meth:`_conclude` call. If set to ``False``, no backends except
| for ``serial`` are supported.
|
| .. note:: If you want to check parallelizability of the whole class, without
| explicitly creating an instance of the class, see
| :attr:`_analysis_algorithm_is_parallelizable`. Note that you
| setting it to other value will break things if the algorithm
| behind the analysis is not trivially parallelizable.
|
|
| Returns
| -------
| bool
| if a given ``AnalysisBase`` subclass instance
| is parallelizable with split-apply-combine, or not
|
|
| .. versionadded:: 2.8.0
|
| ----------------------------------------------------------------------
| Methods inherited from maicos.core.base.ProfileBase:
|
| save(self) -> None
| Save results of analysis to file specified by ``output``.
class DensitySphere(maicos.core.sphere.ProfileSphereBase)
| DensitySphere(
| atomgroup: mda.AtomGroup,
| dens: str = 'mass',
| rmin: float = 0,
| rmax: float | None = None,
| bin_width: float = 1,
| bin_method: str = 'com',
| grouping: str = 'atoms',
| refgroup: mda.AtomGroup | None = None,
| unwrap: bool = True,
| pack: bool = True,
| jitter: float = 0.0,
| concfreq: int = 0,
| output: str = 'density.dat'
| ) -> None
|
| Spherical partial density profiles.
|
| Calculations are carried out for ``mass``
| :math:`(\rm u \cdot Å^{-3})`, ``number`` :math:`(\rm Å^{-3})`, partial ``charge``
| :math:`(\rm e \cdot Å^{-3})` or electron :math:`(\rm e \cdot Å^{-3})` density
| profiles
| along radial axis in spherical coordinate system. The
| origin of the coordinate system defaults to the box center, but can be set to
| dynamically follow the center of mass of a reference group via `refgroup`.
| Cell dimensions are allowed to fluctuate in time.
|
| For grouping with respect to ``molecules``, ``residues`` etc., the corresponding
| centers (i.e., center of mass), taking into account periodic boundary conditions,
| are calculated. For these calculations molecules will be unwrapped/made whole.
| Trajectories containing already whole molecules can be run with ``unwrap=False`` to
| gain a speedup. For grouping with respect to atoms, the ``unwrap`` option is always
| ignored.
|
| For the correlation analysis the 0th bin of the 0th's
| group profile is used. For further information on the correlation analysis please
| refer to :class:`AnalysisBase <maicos.core.base.AnalysisBase>` or the
| :ref:`general-design` section.
|
| Parameters
| ----------
| atomgroup : MDAnalysis.core.groups.AtomGroup
| A :class:`~MDAnalysis.core.groups.AtomGroup` for which the calculations are
| performed.
| dens : {``"mass"``, ``"number"``, ``"charge"``, ``"electron"``}
| density type to be calculated.
| rmin : float
| Minimal radial coordinate relative to the center of mass of the refgroup for
| evaluation (in Å).
| rmax : float
| Maximal radial coordinate relative to the center of mass of the refgroup for
| evaluation (in Å).
|
| If ``rmax=None``, the box extension is taken.
| bin_width : float
| Width of the bins (in Å).
| bin_method : {``"com"``, ``"cog"``, ``"coc"``}
| Method for the position binning.
|
| The possible options are center of mass (``"com"``), center of geometry (``"cog"``),
| and center of charge (``"coc"``).
| grouping : {``"atoms"``, ``"residues"``, ``"segments"``, ``"molecules"``, ``"fragments"``}
| Atom grouping for the calculations.
|
| The possible grouping options are the atom positions (in the case where
| ``grouping="atoms"``) or the center of mass of the specified grouping unit (in the
| case where ``grouping="residues"``, ``"segments"``, ``"molecules"`` or
| ``"fragments"``).
| refgroup : MDAnalysis.core.groups.AtomGroup
| Reference :class:`~MDAnalysis.core.groups.AtomGroup` used for the calculation. If
| ``refgroup`` is provided, the calculation is performed relative to the center of
| mass of the AtomGroup. If ``refgroup`` is :obj:`None` the calculations are performed
| with respect to the center of the (changing) box.
| unwrap : bool
| When :obj:`True`, molecules that are broken due to the periodic boundary conditions
| are made whole.
|
| If the input contains molecules that are already whole, speed up the calculation by
| disabling unwrap. To do so, use the flag ``-no-unwrap`` when using MAICoS from the
| command line, or use ``unwrap=False`` when using MAICoS from the Python interpreter.
|
| Note: Molecules containing virtual sites (e.g. TIP4P water models) are not currently
| supported in MDAnalysis. In this case, you need to provide unwrapped trajectory
| files directly, and disable unwrap. Trajectories can be unwrapped, for example,
| using the ``trjconv`` command of GROMACS.
| pack : bool
| When :obj:`True`, molecules are put back into the unit cell. This is required
| because MAICoS only takes into account molecules that are inside the unit cell.
|
| If the input contains molecules that are already packed, speed up the calculation by
| disabling packing with ``pack=False``.
| jitter : float
| Magnitude of the random noise to add to the atomic positions.
|
| A jitter can be used to stabilize the aliasing effects sometimes appearing when
| histogramming data. The jitter value should be about the precision of the
| trajectory. In that case, using jitter will not alter the results of the histogram.
| If ``jitter = 0.0`` (default), the original atomic positions are kept unchanged.
|
| You can estimate the precision of the positions in your trajectory with
| :func:`maicos.lib.util.trajectory_precision`. Note that if the precision is not the
| same for all frames, the smallest precision should be used.
| concfreq : int
| When concfreq (for conclude frequency) is larger than ``0``, the conclude function
| is called and the output files are written every ``concfreq`` frames.
| output : str
| Output filename.
|
| Attributes
| ----------
| results.bin_pos : numpy.ndarray
| Bin positions (in Å) ranging from ``rmin`` to ``rmax``.
| results.profile : numpy.ndarray
| Calculated profile.
| results.dprofile : numpy.ndarray
| Estimated profile's uncertainity.
|
| Method resolution order:
| DensitySphere
| maicos.core.sphere.ProfileSphereBase
| maicos.core.sphere.SphereBase
| maicos.core.base.AnalysisBase
| maicos.core.base._Runner
| MDAnalysis.analysis.base.AnalysisBase
| maicos.core.base.ProfileBase
| builtins.object
|
| Methods defined here:
|
| __init__(
| self,
| atomgroup: mda.AtomGroup,
| dens: str = 'mass',
| rmin: float = 0,
| rmax: float | None = None,
| bin_width: float = 1,
| bin_method: str = 'com',
| grouping: str = 'atoms',
| refgroup: mda.AtomGroup | None = None,
| unwrap: bool = True,
| pack: bool = True,
| jitter: float = 0.0,
| concfreq: int = 0,
| output: str = 'density.dat'
| ) -> None
| Initialize self. See help(type(self)) for accurate signature.
|
| ----------------------------------------------------------------------
| Methods inherited from maicos.core.base.AnalysisBase:
|
| dump(self, filename: str) -> None
| Save analysis state to an ``.npz`` file.
|
| .. warning::
|
| :meth:`dump` is **not** an archival format. The on-disk layout
| tracks the installed MAICoS and MDAnalysis versions and may stop
| loading after an upgrade. Use it to checkpoint or hand off an
| in-progress analysis, not to preserve results long-term. For
| archival output write summary tables with :meth:`save` or export
| :attr:`results` to a stable format.
|
| Persists all statistical accumulators (``means``, ``sems``, ``sums``,
| ``pop``, ``M2``), the ``results`` and ``_obs`` containers, per-frame
| arrays (``timeseries``, ``frames``, ``times``), metadata, the associated
| Universe and the analysed atomgroup. Restore the analysis with :meth:`load`.
|
| Parameters
| ----------
| filename : str
| Path to the output ``.npz`` file.
|
| run(
| self,
| start: int | None = None,
| stop: int | None = None,
| step: int | None = None,
| frames: int | None = None,
| verbose: bool | None = None,
| progressbar_kwargs: dict | None = None
| ) -> Self
| Iterate over the trajectory.
|
| Parameters
| ----------
| start : int
| start frame of analysis
| stop : int
| stop frame of analysis
| step : int
| number of frames to skip between each analysed frame
| frames : array_like
| array of integers or booleans to slice trajectory; ``frames`` can only be
| used *instead* of ``start``, ``stop``, and ``step``. Setting *both*
| ``frames`` and at least one of ``start``, ``stop``, ``step`` to a
| non-default value will raise a :exc:`ValueError`.
| verbose : bool
| Turn on verbosity
| progressbar_kwargs : dict
| ProgressBar keywords with custom parameters regarding progress bar position,
| etc; see :class:`MDAnalysis.lib.log.ProgressBar` for full list.
|
| Returns
| -------
| self : object
| analysis object
|
| savetxt(self, fname: str, X: np.ndarray, columns: list[str] | None = None) -> None
| Save to text.
|
| An extension of the numpy savetxt function. Adds the command line input to the
| header and checks for a doubled defined filesuffix.
|
| Return a header for the text file to save the data to. This method builds a
| generic header that can be used by any MAICoS module. It is called by the save
| method of each module.
|
| The information it collects is:
| - timestamp of the analysis
| - name of the module
| - version of MAICoS that was used
| - command line arguments that were used to run the module
| - module call including the default arguments
| - number of frames that were analyzed
| - atomgroup that was analyzed
| - output messages from modules and base classes (if they exist)
|
| ----------------------------------------------------------------------
| Class methods inherited from maicos.core.base.AnalysisBase:
|
| load(filename: str) -> Self
| Restore an analysis instance from a file created by :meth:`dump`.
|
| Returns a new instance of ``cls`` with all statistical accumulators,
| per-frame arrays, metadata, and the analysed atomgroup populated from
| the file. The rebuilt :class:`MDAnalysis.Universe` carries only the
| topology — no trajectory — so calling :meth:`run` on the returned
| instance raises :class:`RuntimeError`. Use the loaded instance to
| inspect :attr:`results` or to call :meth:`save`.
|
| Parameters
| ----------
| filename : str
| Path to the ``.npz`` file written by :meth:`dump`.
|
| Returns
| -------
| Self
| A new instance of ``cls`` with state restored from ``filename``.
|
| ----------------------------------------------------------------------
| Readonly properties inherited from maicos.core.base.AnalysisBase:
|
| box_center
| Center of the simulation cell.
|
| box_lengths
| Lengths of the simulation cell vectors.
|
| ----------------------------------------------------------------------
| Data descriptors inherited from maicos.core.base._Runner:
|
| __dict__
| dictionary for instance variables
|
| __weakref__
| list of weak references to the object
|
| ----------------------------------------------------------------------
| Class methods inherited from MDAnalysis.analysis.base.AnalysisBase:
|
| get_supported_backends()
| Tuple with backends supported by the core library for a given class.
| User can pass either one of these values as ``backend=...`` to
| :meth:`run()` method, or a custom object that has ``apply`` method
| (see documentation for :meth:`run()`):
|
| - 'serial': no parallelization
| - 'multiprocessing': parallelization using `multiprocessing.Pool`
| - 'dask': parallelization using `dask.delayed.compute()`. Requires
| installation of `mdanalysis[dask]`
|
| If you want to add your own backend to an existing class, pass a
| :class:`backends.BackendBase` subclass (see its documentation to learn
| how to implement it properly), and specify ``unsupported_backend=True``.
|
| Returns
| -------
| tuple
| names of built-in backends that can be used in :meth:`run(backend=...)`
|
|
| .. versionadded:: 2.8.0
|
| ----------------------------------------------------------------------
| Readonly properties inherited from MDAnalysis.analysis.base.AnalysisBase:
|
| parallelizable
| Boolean mark showing that a given class can be parallelizable with
| split-apply-combine procedure. Namely, if we can safely distribute
| :meth:`_single_frame` to multiple workers and then combine them with a
| proper :meth:`_conclude` call. If set to ``False``, no backends except
| for ``serial`` are supported.
|
| .. note:: If you want to check parallelizability of the whole class, without
| explicitly creating an instance of the class, see
| :attr:`_analysis_algorithm_is_parallelizable`. Note that you
| setting it to other value will break things if the algorithm
| behind the analysis is not trivially parallelizable.
|
|
| Returns
| -------
| bool
| if a given ``AnalysisBase`` subclass instance
| is parallelizable with split-apply-combine, or not
|
|
| .. versionadded:: 2.8.0
|
| ----------------------------------------------------------------------
| Methods inherited from maicos.core.base.ProfileBase:
|
| save(self) -> None
| Save results of analysis to file specified by ``output``.
class DielectricCylinder(maicos.core.cylinder.CylinderBase)
| DielectricCylinder(
| atomgroup: mda.AtomGroup,
| temperature: float = 300,
| vcutwidth: float = 0.1,
| single: bool = False,
| dim: int = 2,
| zmin: float | None = None,
| zmax: float | None = None,
| rmin: float = 0,
| rmax: float | None = None,
| bin_width: float = 0.1,
| refgroup: mda.AtomGroup | None = None,
| unwrap: bool = True,
| pack: bool = True,
| jitter: float = 0.0,
| concfreq: int = 0,
| output_prefix: str = 'eps_cyl'
| ) -> None
|
| Cylindrical dielectric profiles.
|
| Computes the axial :math:`\varepsilon_z(r)`, azimuthal :math:`\varepsilon_{\phi}(r)`
| and inverse radial :math:`\varepsilon_r^{-1}(r)` components of the cylindrical
| dielectric tensor :math:`\varepsilon`.
| The components are binned along the radial direction of the
| cylinder. The :math:`z`-axis of the cylinder is pointing in the direction given by
| the ``dim`` parameter. The center of the cylinder is either located at the center of
| the simulation box (default) or at the center of mass of the ``refgroup``, if
| provided.
|
| For usage please refer to the
| :ref:`sphx_glr_generated_examples_dielectrics_dielectric-profiles.py` example and
| for details on the theory see :ref:`dielectric-explanations`.
|
| For correlation analysis, the component along the :math:`z`-axis is used.
| For further information on the correlation analysis please
| refer to :class:`AnalysisBase <maicos.core.base.AnalysisBase>` or the
| :ref:`general-design` section.
|
| Also, please read and cite :footcite:p:`locheGiantaxialDielectric2019`.
|
| Parameters
| ----------
| atomgroup : MDAnalysis.core.groups.AtomGroup
| A :class:`~MDAnalysis.core.groups.AtomGroup` for which the calculations are
| performed.
| temperature : float
| Reference temperature (K)
| vcutwidth : float
| Spacing of virtual cuts (bins) along the parallel directions.
| single : bool
| For a single chain of molecules the average of :math:`M` is zero. This flag sets
| :math:`\langle M \rangle = 0`.
| dim : {0, 1, 2}
| Dimension for binning (``x=0``, ``y=1``, ``z=1``).
| zmin : float
| Minimal coordinate for evaluation (in Å) with respect to the center of mass of
| the refgroup.
| If ``zmin=None``, all coordinates down to the lower cell boundary are taken into
| account.
| zmax : float
| Maximal coordinate for evaluation (in Å) with respect to the center of mass of the
| refgroup.
| If ``zmax = None``, all coordinates up to the upper cell boundary are taken into
| account.
| rmin : float
| Minimal radial coordinate relative to the center of mass of the refgroup for
| evaluation (in Å).
| rmax : float
| Maximal radial coordinate relative to the center of mass of the refgroup for
| evaluation (in Å).
|
| If ``rmax=None``, the box extension is taken.
| bin_width : float
| Width of the bins (in Å).
| refgroup : MDAnalysis.core.groups.AtomGroup
| Reference :class:`~MDAnalysis.core.groups.AtomGroup` used for the calculation. If
| ``refgroup`` is provided, the calculation is performed relative to the center of
| mass of the AtomGroup. If ``refgroup`` is :obj:`None` the calculations are performed
| with respect to the center of the (changing) box.
| unwrap : bool
| When :obj:`True`, molecules that are broken due to the periodic boundary conditions
| are made whole.
|
| If the input contains molecules that are already whole, speed up the calculation by
| disabling unwrap. To do so, use the flag ``-no-unwrap`` when using MAICoS from the
| command line, or use ``unwrap=False`` when using MAICoS from the Python interpreter.
|
| Note: Molecules containing virtual sites (e.g. TIP4P water models) are not currently
| supported in MDAnalysis. In this case, you need to provide unwrapped trajectory
| files directly, and disable unwrap. Trajectories can be unwrapped, for example,
| using the ``trjconv`` command of GROMACS.
| pack : bool
| When :obj:`True`, molecules are put back into the unit cell. This is required
| because MAICoS only takes into account molecules that are inside the unit cell.
|
| If the input contains molecules that are already packed, speed up the calculation by
| disabling packing with ``pack=False``.
| jitter : float
| Magnitude of the random noise to add to the atomic positions.
|
| A jitter can be used to stabilize the aliasing effects sometimes appearing when
| histogramming data. The jitter value should be about the precision of the
| trajectory. In that case, using jitter will not alter the results of the histogram.
| If ``jitter = 0.0`` (default), the original atomic positions are kept unchanged.
|
| You can estimate the precision of the positions in your trajectory with
| :func:`maicos.lib.util.trajectory_precision`. Note that if the precision is not the
| same for all frames, the smallest precision should be used.
| concfreq : int
| When concfreq (for conclude frequency) is larger than ``0``, the conclude function
| is called and the output files are written every ``concfreq`` frames.
| output_prefix : str
| Prefix for output files.
|
| Attributes
| ----------
| results.bin_pos : numpy.ndarray
| Bin positions (in Å) ranging from ``rmin`` to ``rmax``.
| results.eps_z : numpy.ndarray
| Reduced axial dielectric profile :math:`(\varepsilon_z(r) - 1)` of the
| selected atomgroup
| results.deps_z : numpy.ndarray
| Estimated uncertainty of axial dielectric profile
| results.eps_r : numpy.ndarray
| Reduced inverse radial dielectric profile
| :math:`(\varepsilon^{-1}_r(r) - 1)`
| results.deps_r : numpy.ndarray
| Estimated uncertainty of inverse radial dielectric profile
| results.eps_phi : numpy.ndarray
| Reduced azimuthal dielectric profile
| :math:`(\varepsilon_{\phi}(r) - 1)`
| results.deps_phi : numpy.ndarray
| Estimated uncertainty of azimuthal dielectric profile.
|
| Method resolution order:
| DielectricCylinder
| maicos.core.cylinder.CylinderBase
| maicos.core.planar.PlanarBase
| maicos.core.base.AnalysisBase
| maicos.core.base._Runner
| MDAnalysis.analysis.base.AnalysisBase
| builtins.object
|
| Methods defined here:
|
| __init__(
| self,
| atomgroup: mda.AtomGroup,
| temperature: float = 300,
| vcutwidth: float = 0.1,
| single: bool = False,
| dim: int = 2,
| zmin: float | None = None,
| zmax: float | None = None,
| rmin: float = 0,
| rmax: float | None = None,
| bin_width: float = 0.1,
| refgroup: mda.AtomGroup | None = None,
| unwrap: bool = True,
| pack: bool = True,
| jitter: float = 0.0,
| concfreq: int = 0,
| output_prefix: str = 'eps_cyl'
| ) -> None
| Initialize self. See help(type(self)) for accurate signature.
|
| save(self) -> None
| Save results of analysis to files specified by
| ``output_prefix``.
|
| ----------------------------------------------------------------------
| Readonly properties inherited from maicos.core.planar.PlanarBase:
|
| odims
| Other dimensions perpendicular to dim i.e. (0,2) if dim = 1.
|
| ----------------------------------------------------------------------
| Methods inherited from maicos.core.base.AnalysisBase:
|
| dump(self, filename: str) -> None
| Save analysis state to an ``.npz`` file.
|
| .. warning::
|
| :meth:`dump` is **not** an archival format. The on-disk layout
| tracks the installed MAICoS and MDAnalysis versions and may stop
| loading after an upgrade. Use it to checkpoint or hand off an
| in-progress analysis, not to preserve results long-term. For
| archival output write summary tables with :meth:`save` or export
| :attr:`results` to a stable format.
|
| Persists all statistical accumulators (``means``, ``sems``, ``sums``,
| ``pop``, ``M2``), the ``results`` and ``_obs`` containers, per-frame
| arrays (``timeseries``, ``frames``, ``times``), metadata, the associated
| Universe and the analysed atomgroup. Restore the analysis with :meth:`load`.
|
| Parameters
| ----------
| filename : str
| Path to the output ``.npz`` file.
|
| run(
| self,
| start: int | None = None,
| stop: int | None = None,
| step: int | None = None,
| frames: int | None = None,
| verbose: bool | None = None,
| progressbar_kwargs: dict | None = None
| ) -> Self
| Iterate over the trajectory.
|
| Parameters
| ----------
| start : int
| start frame of analysis
| stop : int
| stop frame of analysis
| step : int
| number of frames to skip between each analysed frame
| frames : array_like
| array of integers or booleans to slice trajectory; ``frames`` can only be
| used *instead* of ``start``, ``stop``, and ``step``. Setting *both*
| ``frames`` and at least one of ``start``, ``stop``, ``step`` to a
| non-default value will raise a :exc:`ValueError`.
| verbose : bool
| Turn on verbosity
| progressbar_kwargs : dict
| ProgressBar keywords with custom parameters regarding progress bar position,
| etc; see :class:`MDAnalysis.lib.log.ProgressBar` for full list.
|
| Returns
| -------
| self : object
| analysis object
|
| savetxt(self, fname: str, X: np.ndarray, columns: list[str] | None = None) -> None
| Save to text.
|
| An extension of the numpy savetxt function. Adds the command line input to the
| header and checks for a doubled defined filesuffix.
|
| Return a header for the text file to save the data to. This method builds a
| generic header that can be used by any MAICoS module. It is called by the save
| method of each module.
|
| The information it collects is:
| - timestamp of the analysis
| - name of the module
| - version of MAICoS that was used
| - command line arguments that were used to run the module
| - module call including the default arguments
| - number of frames that were analyzed
| - atomgroup that was analyzed
| - output messages from modules and base classes (if they exist)
|
| ----------------------------------------------------------------------
| Class methods inherited from maicos.core.base.AnalysisBase:
|
| load(filename: str) -> Self
| Restore an analysis instance from a file created by :meth:`dump`.
|
| Returns a new instance of ``cls`` with all statistical accumulators,
| per-frame arrays, metadata, and the analysed atomgroup populated from
| the file. The rebuilt :class:`MDAnalysis.Universe` carries only the
| topology — no trajectory — so calling :meth:`run` on the returned
| instance raises :class:`RuntimeError`. Use the loaded instance to
| inspect :attr:`results` or to call :meth:`save`.
|
| Parameters
| ----------
| filename : str
| Path to the ``.npz`` file written by :meth:`dump`.
|
| Returns
| -------
| Self
| A new instance of ``cls`` with state restored from ``filename``.
|
| ----------------------------------------------------------------------
| Readonly properties inherited from maicos.core.base.AnalysisBase:
|
| box_center
| Center of the simulation cell.
|
| box_lengths
| Lengths of the simulation cell vectors.
|
| ----------------------------------------------------------------------
| Data descriptors inherited from maicos.core.base._Runner:
|
| __dict__
| dictionary for instance variables
|
| __weakref__
| list of weak references to the object
|
| ----------------------------------------------------------------------
| Class methods inherited from MDAnalysis.analysis.base.AnalysisBase:
|
| get_supported_backends()
| Tuple with backends supported by the core library for a given class.
| User can pass either one of these values as ``backend=...`` to
| :meth:`run()` method, or a custom object that has ``apply`` method
| (see documentation for :meth:`run()`):
|
| - 'serial': no parallelization
| - 'multiprocessing': parallelization using `multiprocessing.Pool`
| - 'dask': parallelization using `dask.delayed.compute()`. Requires
| installation of `mdanalysis[dask]`
|
| If you want to add your own backend to an existing class, pass a
| :class:`backends.BackendBase` subclass (see its documentation to learn
| how to implement it properly), and specify ``unsupported_backend=True``.
|
| Returns
| -------
| tuple
| names of built-in backends that can be used in :meth:`run(backend=...)`
|
|
| .. versionadded:: 2.8.0
|
| ----------------------------------------------------------------------
| Readonly properties inherited from MDAnalysis.analysis.base.AnalysisBase:
|
| parallelizable
| Boolean mark showing that a given class can be parallelizable with
| split-apply-combine procedure. Namely, if we can safely distribute
| :meth:`_single_frame` to multiple workers and then combine them with a
| proper :meth:`_conclude` call. If set to ``False``, no backends except
| for ``serial`` are supported.
|
| .. note:: If you want to check parallelizability of the whole class, without
| explicitly creating an instance of the class, see
| :attr:`_analysis_algorithm_is_parallelizable`. Note that you
| setting it to other value will break things if the algorithm
| behind the analysis is not trivially parallelizable.
|
|
| Returns
| -------
| bool
| if a given ``AnalysisBase`` subclass instance
| is parallelizable with split-apply-combine, or not
|
|
| .. versionadded:: 2.8.0
class DielectricPlanar(maicos.core.planar.PlanarBase)
| DielectricPlanar(
| atomgroup: mda.AtomGroup,
| temperature: float = 300,
| vcutwidth: float = 0.1,
| is_3d: bool = False,
| dim: int = 2,
| zmin: float | None = None,
| zmax: float | None = None,
| bin_width: float = 0.5,
| sym: bool = False,
| refgroup: mda.AtomGroup | None = None,
| unwrap: bool = True,
| pack: bool = True,
| jitter: float = 0.0,
| concfreq: int = 0,
| output_prefix: str = 'eps'
| ) -> None
|
| Planar dielectric profiles.
|
| Computes the parallel :math:`\varepsilon_\parallel(z)` and inverse perpendicular
| (:math:`\varepsilon_\perp^{-1}(r)`) components of the planar dielectric tensor
| :math:`\varepsilon`. The components are binned along the cartesian :math:`z`
| direction yielding the component normal to the surface and defined by the ``dim``
| parameter.
|
| For usage please refer to the
| :ref:`sphx_glr_generated_examples_dielectrics_dielectric-profiles.py` example and
| for details on the theory see :ref:`dielectric-explanations`.
|
| For correlation analysis, the norm of the parallel total dipole moment is used.
| For further information on the correlation analysis please
| refer to :class:`AnalysisBase <maicos.core.base.AnalysisBase>` or the
| :ref:`general-design` section.
|
| Also, please read and cite
| :footcite:t:`schlaichWaterDielectricEffects2016` and Refs.
| :footcite:p:`locheUniversalNonuniversalAspects2020`,
| :footcite:p:`bonthuisProfileStaticPermittivity2012`.
|
| Parameters
| ----------
| atomgroup : MDAnalysis.core.groups.AtomGroup
| A :class:`~MDAnalysis.core.groups.AtomGroup` for which the calculations are
| performed.
| temperature : float
| Reference temperature (K)
| vcutwidth : float
| Spacing of virtual cuts (bins) along the parallel directions.
| is_3d : bool
| Use 3d-periodic boundary conditions, i.e., include the dipole correction for
| the interaction between periodic images
| :footcite:p:`sternCalculationDielectricPermittivity2003`.
| dim : {0, 1, 2}
| Dimension for binning (``x=0``, ``y=1``, ``z=1``).
| zmin : float
| Minimal coordinate for evaluation (in Å) with respect to the center of mass of
| the refgroup.
| If ``zmin=None``, all coordinates down to the lower cell boundary are taken into
| account.
| zmax : float
| Maximal coordinate for evaluation (in Å) with respect to the center of mass of the
| refgroup.
| If ``zmax = None``, all coordinates up to the upper cell boundary are taken into
| account.
| bin_width : float
| Width of the bins (in Å).
| sym : bool
| Symmetrize the profile. Only works in combination with ``refgroup``.
| refgroup : MDAnalysis.core.groups.AtomGroup
| Reference :class:`~MDAnalysis.core.groups.AtomGroup` used for the calculation. If
| ``refgroup`` is provided, the calculation is performed relative to the center of
| mass of the AtomGroup. If ``refgroup`` is :obj:`None` the calculations are performed
| with respect to the center of the (changing) box.
| unwrap : bool
| When :obj:`True`, molecules that are broken due to the periodic boundary conditions
| are made whole.
|
| If the input contains molecules that are already whole, speed up the calculation by
| disabling unwrap. To do so, use the flag ``-no-unwrap`` when using MAICoS from the
| command line, or use ``unwrap=False`` when using MAICoS from the Python interpreter.
|
| Note: Molecules containing virtual sites (e.g. TIP4P water models) are not currently
| supported in MDAnalysis. In this case, you need to provide unwrapped trajectory
| files directly, and disable unwrap. Trajectories can be unwrapped, for example,
| using the ``trjconv`` command of GROMACS.
| pack : bool
| When :obj:`True`, molecules are put back into the unit cell. This is required
| because MAICoS only takes into account molecules that are inside the unit cell.
|
| If the input contains molecules that are already packed, speed up the calculation by
| disabling packing with ``pack=False``.
| jitter : float
| Magnitude of the random noise to add to the atomic positions.
|
| A jitter can be used to stabilize the aliasing effects sometimes appearing when
| histogramming data. The jitter value should be about the precision of the
| trajectory. In that case, using jitter will not alter the results of the histogram.
| If ``jitter = 0.0`` (default), the original atomic positions are kept unchanged.
|
| You can estimate the precision of the positions in your trajectory with
| :func:`maicos.lib.util.trajectory_precision`. Note that if the precision is not the
| same for all frames, the smallest precision should be used.
| concfreq : int
| When concfreq (for conclude frequency) is larger than ``0``, the conclude function
| is called and the output files are written every ``concfreq`` frames.
| output_prefix : str
| Prefix for output files.
|
| Attributes
| ----------
| results.bin_pos : numpy.ndarray
| Bin positions (in Å) ranging from ``zmin`` to ``zmax``.
| results.eps_par : numpy.ndarray
| Reduced parallel dielectric profile
| :math:`(\varepsilon_\parallel(z) - 1)` of the selected AtomGroup
| results.deps_par : numpy.ndarray
| Uncertainty of parallel dielectric profile
| results.eps_par_self : numpy.ndarray
| Reduced self contribution of parallel dielectric profile
| :math:`(\varepsilon_{\parallel,\mathrm{self}}(z) - 1)`
| results.eps_par_coll : numpy.ndarray
| Reduced collective contribution of parallel dielectric profile
| :math:`(\varepsilon_{\parallel,\mathrm{coll}}(z) - 1)`
| results.eps_perp : numpy.ndarray
| Reduced inverse perpendicular dielectric profile
| :math:`(\varepsilon^{-1}_\perp(z) - 1)`
| results.deps_perp : numpy.ndarray
| Uncertainty of inverse perpendicular dielectric profile
| results.eps_perp_self : numpy.ndarray
| Reduced self contribution of the inverse perpendicular dielectric
| profile :math:`(\varepsilon^{-1}_{\perp,\mathrm{self}}(z) - 1)`
| results.eps_perp_coll : numpy.ndarray
| Reduced collective contribution of the inverse perpendicular dielectric profile
| :math:`(\varepsilon^{-1}_{\perp,\mathrm{coll}}(z) - 1)`
|
| Method resolution order:
| DielectricPlanar
| maicos.core.planar.PlanarBase
| maicos.core.base.AnalysisBase
| maicos.core.base._Runner
| MDAnalysis.analysis.base.AnalysisBase
| builtins.object
|
| Methods defined here:
|
| __init__(
| self,
| atomgroup: mda.AtomGroup,
| temperature: float = 300,
| vcutwidth: float = 0.1,
| is_3d: bool = False,
| dim: int = 2,
| zmin: float | None = None,
| zmax: float | None = None,
| bin_width: float = 0.5,
| sym: bool = False,
| refgroup: mda.AtomGroup | None = None,
| unwrap: bool = True,
| pack: bool = True,
| jitter: float = 0.0,
| concfreq: int = 0,
| output_prefix: str = 'eps'
| ) -> None
| Initialize self. See help(type(self)) for accurate signature.
|
| save(self) -> None
| Save results of analysis to files specified by
| ``output_prefix``.
|
| ----------------------------------------------------------------------
| Readonly properties inherited from maicos.core.planar.PlanarBase:
|
| odims
| Other dimensions perpendicular to dim i.e. (0,2) if dim = 1.
|
| ----------------------------------------------------------------------
| Methods inherited from maicos.core.base.AnalysisBase:
|
| dump(self, filename: str) -> None
| Save analysis state to an ``.npz`` file.
|
| .. warning::
|
| :meth:`dump` is **not** an archival format. The on-disk layout
| tracks the installed MAICoS and MDAnalysis versions and may stop
| loading after an upgrade. Use it to checkpoint or hand off an
| in-progress analysis, not to preserve results long-term. For
| archival output write summary tables with :meth:`save` or export
| :attr:`results` to a stable format.
|
| Persists all statistical accumulators (``means``, ``sems``, ``sums``,
| ``pop``, ``M2``), the ``results`` and ``_obs`` containers, per-frame
| arrays (``timeseries``, ``frames``, ``times``), metadata, the associated
| Universe and the analysed atomgroup. Restore the analysis with :meth:`load`.
|
| Parameters
| ----------
| filename : str
| Path to the output ``.npz`` file.
|
| run(
| self,
| start: int | None = None,
| stop: int | None = None,
| step: int | None = None,
| frames: int | None = None,
| verbose: bool | None = None,
| progressbar_kwargs: dict | None = None
| ) -> Self
| Iterate over the trajectory.
|
| Parameters
| ----------
| start : int
| start frame of analysis
| stop : int
| stop frame of analysis
| step : int
| number of frames to skip between each analysed frame
| frames : array_like
| array of integers or booleans to slice trajectory; ``frames`` can only be
| used *instead* of ``start``, ``stop``, and ``step``. Setting *both*
| ``frames`` and at least one of ``start``, ``stop``, ``step`` to a
| non-default value will raise a :exc:`ValueError`.
| verbose : bool
| Turn on verbosity
| progressbar_kwargs : dict
| ProgressBar keywords with custom parameters regarding progress bar position,
| etc; see :class:`MDAnalysis.lib.log.ProgressBar` for full list.
|
| Returns
| -------
| self : object
| analysis object
|
| savetxt(self, fname: str, X: np.ndarray, columns: list[str] | None = None) -> None
| Save to text.
|
| An extension of the numpy savetxt function. Adds the command line input to the
| header and checks for a doubled defined filesuffix.
|
| Return a header for the text file to save the data to. This method builds a
| generic header that can be used by any MAICoS module. It is called by the save
| method of each module.
|
| The information it collects is:
| - timestamp of the analysis
| - name of the module
| - version of MAICoS that was used
| - command line arguments that were used to run the module
| - module call including the default arguments
| - number of frames that were analyzed
| - atomgroup that was analyzed
| - output messages from modules and base classes (if they exist)
|
| ----------------------------------------------------------------------
| Class methods inherited from maicos.core.base.AnalysisBase:
|
| load(filename: str) -> Self
| Restore an analysis instance from a file created by :meth:`dump`.
|
| Returns a new instance of ``cls`` with all statistical accumulators,
| per-frame arrays, metadata, and the analysed atomgroup populated from
| the file. The rebuilt :class:`MDAnalysis.Universe` carries only the
| topology — no trajectory — so calling :meth:`run` on the returned
| instance raises :class:`RuntimeError`. Use the loaded instance to
| inspect :attr:`results` or to call :meth:`save`.
|
| Parameters
| ----------
| filename : str
| Path to the ``.npz`` file written by :meth:`dump`.
|
| Returns
| -------
| Self
| A new instance of ``cls`` with state restored from ``filename``.
|
| ----------------------------------------------------------------------
| Readonly properties inherited from maicos.core.base.AnalysisBase:
|
| box_center
| Center of the simulation cell.
|
| box_lengths
| Lengths of the simulation cell vectors.
|
| ----------------------------------------------------------------------
| Data descriptors inherited from maicos.core.base._Runner:
|
| __dict__
| dictionary for instance variables
|
| __weakref__
| list of weak references to the object
|
| ----------------------------------------------------------------------
| Class methods inherited from MDAnalysis.analysis.base.AnalysisBase:
|
| get_supported_backends()
| Tuple with backends supported by the core library for a given class.
| User can pass either one of these values as ``backend=...`` to
| :meth:`run()` method, or a custom object that has ``apply`` method
| (see documentation for :meth:`run()`):
|
| - 'serial': no parallelization
| - 'multiprocessing': parallelization using `multiprocessing.Pool`
| - 'dask': parallelization using `dask.delayed.compute()`. Requires
| installation of `mdanalysis[dask]`
|
| If you want to add your own backend to an existing class, pass a
| :class:`backends.BackendBase` subclass (see its documentation to learn
| how to implement it properly), and specify ``unsupported_backend=True``.
|
| Returns
| -------
| tuple
| names of built-in backends that can be used in :meth:`run(backend=...)`
|
|
| .. versionadded:: 2.8.0
|
| ----------------------------------------------------------------------
| Readonly properties inherited from MDAnalysis.analysis.base.AnalysisBase:
|
| parallelizable
| Boolean mark showing that a given class can be parallelizable with
| split-apply-combine procedure. Namely, if we can safely distribute
| :meth:`_single_frame` to multiple workers and then combine them with a
| proper :meth:`_conclude` call. If set to ``False``, no backends except
| for ``serial`` are supported.
|
| .. note:: If you want to check parallelizability of the whole class, without
| explicitly creating an instance of the class, see
| :attr:`_analysis_algorithm_is_parallelizable`. Note that you
| setting it to other value will break things if the algorithm
| behind the analysis is not trivially parallelizable.
|
|
| Returns
| -------
| bool
| if a given ``AnalysisBase`` subclass instance
| is parallelizable with split-apply-combine, or not
|
|
| .. versionadded:: 2.8.0
class DielectricSpectrum(builtins.object)
| DielectricSpectrum(
| atomgroup: mda.AtomGroup,
| temperature: float = 300,
| segs: int = 20,
| df: float | None = None,
| bins: int = 200,
| binafter: float = 20,
| nobin: bool = False,
| refgroup: mda.AtomGroup | None = None,
| unwrap: bool = True,
| pack: bool = True,
| jitter: float = 0.0,
| concfreq: int = 0,
| output_prefix: str = ''
| ) -> None
|
| Class has been moved to the ``spectrakit`` package.
|
| Methods defined here:
|
| __init__(
| self,
| atomgroup: mda.AtomGroup,
| temperature: float = 300,
| segs: int = 20,
| df: float | None = None,
| bins: int = 200,
| binafter: float = 20,
| nobin: bool = False,
| refgroup: mda.AtomGroup | None = None,
| unwrap: bool = True,
| pack: bool = True,
| jitter: float = 0.0,
| concfreq: int = 0,
| output_prefix: str = ''
| ) -> None
| Initialize self. See help(type(self)) for accurate signature.
|
| ----------------------------------------------------------------------
| Data descriptors defined here:
|
| __dict__
| dictionary for instance variables
|
| __weakref__
| list of weak references to the object
class DielectricSphere(maicos.core.sphere.SphereBase)
| DielectricSphere(
| atomgroup: mda.AtomGroup,
| temperature: float = 300,
| rmin: float = 0,
| rmax: float | None = None,
| bin_width: float = 0.1,
| refgroup: mda.AtomGroup | None = None,
| unwrap: bool = True,
| pack: bool = True,
| jitter: float = 0.0,
| concfreq: int = 0,
| output_prefix: str = 'eps_sph'
| ) -> None
|
| Spherical dielectric profiles.
|
| Computes the inverse radial :math:`\varepsilon_r^{-1}(r)` component of the
| spherical dielectric tensor :math:`\varepsilon`. The center of the sphere is either
| located at the center of the simulation box (default) or at the center of mass of
| the ``refgroup``, if provided.
|
| For usage, please refer to
| :ref:`sphx_glr_generated_examples_dielectrics_dielectric-profiles.py` and for
| details on the theory see :ref:`dielectric-explanations`.
|
| For correlation analysis, the radial (:math:`r`) component is used.
| For further information on the correlation analysis please
| refer to :class:`AnalysisBase <maicos.core.base.AnalysisBase>` or the
| :ref:`general-design` section.
|
| Also, please read and cite :footcite:p:`schaafDielectricResponseWater2015`.
|
| Parameters
| ----------
| atomgroup : MDAnalysis.core.groups.AtomGroup
| A :class:`~MDAnalysis.core.groups.AtomGroup` for which the calculations are
| performed.
| temperature : float
| Reference temperature (K)
| rmin : float
| Minimal radial coordinate relative to the center of mass of the refgroup for
| evaluation (in Å).
| rmax : float
| Maximal radial coordinate relative to the center of mass of the refgroup for
| evaluation (in Å).
|
| If ``rmax=None``, the box extension is taken.
| bin_width : float
| Width of the bins (in Å).
| refgroup : MDAnalysis.core.groups.AtomGroup
| Reference :class:`~MDAnalysis.core.groups.AtomGroup` used for the calculation. If
| ``refgroup`` is provided, the calculation is performed relative to the center of
| mass of the AtomGroup. If ``refgroup`` is :obj:`None` the calculations are performed
| with respect to the center of the (changing) box.
| unwrap : bool
| When :obj:`True`, molecules that are broken due to the periodic boundary conditions
| are made whole.
|
| If the input contains molecules that are already whole, speed up the calculation by
| disabling unwrap. To do so, use the flag ``-no-unwrap`` when using MAICoS from the
| command line, or use ``unwrap=False`` when using MAICoS from the Python interpreter.
|
| Note: Molecules containing virtual sites (e.g. TIP4P water models) are not currently
| supported in MDAnalysis. In this case, you need to provide unwrapped trajectory
| files directly, and disable unwrap. Trajectories can be unwrapped, for example,
| using the ``trjconv`` command of GROMACS.
| pack : bool
| When :obj:`True`, molecules are put back into the unit cell. This is required
| because MAICoS only takes into account molecules that are inside the unit cell.
|
| If the input contains molecules that are already packed, speed up the calculation by
| disabling packing with ``pack=False``.
| jitter : float
| Magnitude of the random noise to add to the atomic positions.
|
| A jitter can be used to stabilize the aliasing effects sometimes appearing when
| histogramming data. The jitter value should be about the precision of the
| trajectory. In that case, using jitter will not alter the results of the histogram.
| If ``jitter = 0.0`` (default), the original atomic positions are kept unchanged.
|
| You can estimate the precision of the positions in your trajectory with
| :func:`maicos.lib.util.trajectory_precision`. Note that if the precision is not the
| same for all frames, the smallest precision should be used.
| concfreq : int
| When concfreq (for conclude frequency) is larger than ``0``, the conclude function
| is called and the output files are written every ``concfreq`` frames.
| output_prefix : str
| Prefix for output files.
|
| Attributes
| ----------
| results.bin_pos : numpy.ndarray
| Bin positions (in Å) ranging from ``rmin`` to ``rmax``.
| results.eps_rad : numpy.ndarray
| Reduced inverse radial dielectric profile (:math:`\varepsilon^{-1}_r(r) - 1)`
| results.deps_rad : numpy.ndarray
| Uncertainty of inverse radial dielectric profile
|
| Method resolution order:
| DielectricSphere
| maicos.core.sphere.SphereBase
| maicos.core.base.AnalysisBase
| maicos.core.base._Runner
| MDAnalysis.analysis.base.AnalysisBase
| builtins.object
|
| Methods defined here:
|
| __init__(
| self,
| atomgroup: mda.AtomGroup,
| temperature: float = 300,
| rmin: float = 0,
| rmax: float | None = None,
| bin_width: float = 0.1,
| refgroup: mda.AtomGroup | None = None,
| unwrap: bool = True,
| pack: bool = True,
| jitter: float = 0.0,
| concfreq: int = 0,
| output_prefix: str = 'eps_sph'
| ) -> None
| Initialize self. See help(type(self)) for accurate signature.
|
| save(self) -> None
| Save results of analysis to files specified by
| ``output_prefix``.
|
| ----------------------------------------------------------------------
| Methods inherited from maicos.core.base.AnalysisBase:
|
| dump(self, filename: str) -> None
| Save analysis state to an ``.npz`` file.
|
| .. warning::
|
| :meth:`dump` is **not** an archival format. The on-disk layout
| tracks the installed MAICoS and MDAnalysis versions and may stop
| loading after an upgrade. Use it to checkpoint or hand off an
| in-progress analysis, not to preserve results long-term. For
| archival output write summary tables with :meth:`save` or export
| :attr:`results` to a stable format.
|
| Persists all statistical accumulators (``means``, ``sems``, ``sums``,
| ``pop``, ``M2``), the ``results`` and ``_obs`` containers, per-frame
| arrays (``timeseries``, ``frames``, ``times``), metadata, the associated
| Universe and the analysed atomgroup. Restore the analysis with :meth:`load`.
|
| Parameters
| ----------
| filename : str
| Path to the output ``.npz`` file.
|
| run(
| self,
| start: int | None = None,
| stop: int | None = None,
| step: int | None = None,
| frames: int | None = None,
| verbose: bool | None = None,
| progressbar_kwargs: dict | None = None
| ) -> Self
| Iterate over the trajectory.
|
| Parameters
| ----------
| start : int
| start frame of analysis
| stop : int
| stop frame of analysis
| step : int
| number of frames to skip between each analysed frame
| frames : array_like
| array of integers or booleans to slice trajectory; ``frames`` can only be
| used *instead* of ``start``, ``stop``, and ``step``. Setting *both*
| ``frames`` and at least one of ``start``, ``stop``, ``step`` to a
| non-default value will raise a :exc:`ValueError`.
| verbose : bool
| Turn on verbosity
| progressbar_kwargs : dict
| ProgressBar keywords with custom parameters regarding progress bar position,
| etc; see :class:`MDAnalysis.lib.log.ProgressBar` for full list.
|
| Returns
| -------
| self : object
| analysis object
|
| savetxt(self, fname: str, X: np.ndarray, columns: list[str] | None = None) -> None
| Save to text.
|
| An extension of the numpy savetxt function. Adds the command line input to the
| header and checks for a doubled defined filesuffix.
|
| Return a header for the text file to save the data to. This method builds a
| generic header that can be used by any MAICoS module. It is called by the save
| method of each module.
|
| The information it collects is:
| - timestamp of the analysis
| - name of the module
| - version of MAICoS that was used
| - command line arguments that were used to run the module
| - module call including the default arguments
| - number of frames that were analyzed
| - atomgroup that was analyzed
| - output messages from modules and base classes (if they exist)
|
| ----------------------------------------------------------------------
| Class methods inherited from maicos.core.base.AnalysisBase:
|
| load(filename: str) -> Self
| Restore an analysis instance from a file created by :meth:`dump`.
|
| Returns a new instance of ``cls`` with all statistical accumulators,
| per-frame arrays, metadata, and the analysed atomgroup populated from
| the file. The rebuilt :class:`MDAnalysis.Universe` carries only the
| topology — no trajectory — so calling :meth:`run` on the returned
| instance raises :class:`RuntimeError`. Use the loaded instance to
| inspect :attr:`results` or to call :meth:`save`.
|
| Parameters
| ----------
| filename : str
| Path to the ``.npz`` file written by :meth:`dump`.
|
| Returns
| -------
| Self
| A new instance of ``cls`` with state restored from ``filename``.
|
| ----------------------------------------------------------------------
| Readonly properties inherited from maicos.core.base.AnalysisBase:
|
| box_center
| Center of the simulation cell.
|
| box_lengths
| Lengths of the simulation cell vectors.
|
| ----------------------------------------------------------------------
| Data descriptors inherited from maicos.core.base._Runner:
|
| __dict__
| dictionary for instance variables
|
| __weakref__
| list of weak references to the object
|
| ----------------------------------------------------------------------
| Class methods inherited from MDAnalysis.analysis.base.AnalysisBase:
|
| get_supported_backends()
| Tuple with backends supported by the core library for a given class.
| User can pass either one of these values as ``backend=...`` to
| :meth:`run()` method, or a custom object that has ``apply`` method
| (see documentation for :meth:`run()`):
|
| - 'serial': no parallelization
| - 'multiprocessing': parallelization using `multiprocessing.Pool`
| - 'dask': parallelization using `dask.delayed.compute()`. Requires
| installation of `mdanalysis[dask]`
|
| If you want to add your own backend to an existing class, pass a
| :class:`backends.BackendBase` subclass (see its documentation to learn
| how to implement it properly), and specify ``unsupported_backend=True``.
|
| Returns
| -------
| tuple
| names of built-in backends that can be used in :meth:`run(backend=...)`
|
|
| .. versionadded:: 2.8.0
|
| ----------------------------------------------------------------------
| Readonly properties inherited from MDAnalysis.analysis.base.AnalysisBase:
|
| parallelizable
| Boolean mark showing that a given class can be parallelizable with
| split-apply-combine procedure. Namely, if we can safely distribute
| :meth:`_single_frame` to multiple workers and then combine them with a
| proper :meth:`_conclude` call. If set to ``False``, no backends except
| for ``serial`` are supported.
|
| .. note:: If you want to check parallelizability of the whole class, without
| explicitly creating an instance of the class, see
| :attr:`_analysis_algorithm_is_parallelizable`. Note that you
| setting it to other value will break things if the algorithm
| behind the analysis is not trivially parallelizable.
|
|
| Returns
| -------
| bool
| if a given ``AnalysisBase`` subclass instance
| is parallelizable with split-apply-combine, or not
|
|
| .. versionadded:: 2.8.0
class DipoleAngle(builtins.object)
| DipoleAngle(
| atomgroup: mda.AtomGroup,
| pdim: int = 2,
| grouping: str = 'residues',
| refgroup: mda.AtomGroup | None = None,
| unwrap: bool = False,
| pack: bool = True,
| jitter: float = 0.0,
| concfreq: int = 0,
| output: str = 'dipangle.dat'
| ) -> None
|
| Class has been removed and is no longer available.
|
| Methods defined here:
|
| __init__(
| self,
| atomgroup: mda.AtomGroup,
| pdim: int = 2,
| grouping: str = 'residues',
| refgroup: mda.AtomGroup | None = None,
| unwrap: bool = False,
| pack: bool = True,
| jitter: float = 0.0,
| concfreq: int = 0,
| output: str = 'dipangle.dat'
| ) -> None
| Initialize self. See help(type(self)) for accurate signature.
|
| ----------------------------------------------------------------------
| Data descriptors defined here:
|
| __dict__
| dictionary for instance variables
|
| __weakref__
| list of weak references to the object
class DiporderCylinder(maicos.core.cylinder.ProfileCylinderBase)
| DiporderCylinder(
| atomgroup: mda.AtomGroup,
| order_parameter: str = 'P1',
| pdim: str = 'r',
| dim: int = 2,
| zmin: float | None = None,
| zmax: float | None = None,
| rmin: float = 0,
| rmax: float | None = None,
| bin_width: float = 1,
| bin_method: str = 'com',
| grouping: str = 'residues',
| refgroup: mda.AtomGroup | None = None,
| unwrap: bool = True,
| pack: bool = True,
| jitter: float = 0.0,
| concfreq: int = 0,
| output: str = 'diporder_cylinder.dat'
| ) -> None
|
| Cylindrical dipolar order parameters.
|
| Calculations include the first- and second-rank
| orientational order parameters,
| which are the Legendre polynomials :math:`P_1(x) = x` and :math:`P_2(x) = \frac{3
| x^2 - 1}{2}` with :math:`x = \cos(θ[z])`.
|
| For the correlation analysis the 0th bin of the 0th's
| group profile is used. For further information on the correlation analysis please
| refer to :class:`AnalysisBase <maicos.core.base.AnalysisBase>` or the
| :ref:`general-design` section.
|
| Parameters
| ----------
| atomgroup : MDAnalysis.core.groups.AtomGroup
| A :class:`~MDAnalysis.core.groups.AtomGroup` for which the calculations are
| performed.
| order_parameter : {``"P1"``, ``"P2"``}
| Order parameter to be calculated:
| - ``"P1"``: Legendre polynomial :math:`P_1(cos[θ]) = cos[θ]`
| - ``"P2"``: Legendre polynomial
| :math:`P_2(cos[θ]) = \frac{3cos^2[θ] - 1}{2}`
| pdim : {``"r"``, ``"z"``}
| direction of the projection
| dim : {0, 1, 2}
| Dimension for binning (``x=0``, ``y=1``, ``z=1``).
| zmin : float
| Minimal coordinate for evaluation (in Å) with respect to the center of mass of
| the refgroup.
| If ``zmin=None``, all coordinates down to the lower cell boundary are taken into
| account.
| zmax : float
| Maximal coordinate for evaluation (in Å) with respect to the center of mass of the
| refgroup.
| If ``zmax = None``, all coordinates up to the upper cell boundary are taken into
| account.
| rmin : float
| Minimal radial coordinate relative to the center of mass of the refgroup for
| evaluation (in Å).
| rmax : float
| Maximal radial coordinate relative to the center of mass of the refgroup for
| evaluation (in Å).
|
| If ``rmax=None``, the box extension is taken.
| bin_width : float
| Width of the bins (in Å).
| bin_method : {``"com"``, ``"cog"``, ``"coc"``}
| Method for the position binning.
|
| The possible options are center of mass (``"com"``), center of geometry (``"cog"``),
| and center of charge (``"coc"``).
| grouping : {``"atoms"``, ``"residues"``, ``"segments"``, ``"molecules"``, ``"fragments"``}
| Atom grouping for the calculations.
|
| The possible grouping options are the atom positions (in the case where
| ``grouping="atoms"``) or the center of mass of the specified grouping unit (in the
| case where ``grouping="residues"``, ``"segments"``, ``"molecules"`` or
| ``"fragments"``).
| refgroup : MDAnalysis.core.groups.AtomGroup
| Reference :class:`~MDAnalysis.core.groups.AtomGroup` used for the calculation. If
| ``refgroup`` is provided, the calculation is performed relative to the center of
| mass of the AtomGroup. If ``refgroup`` is :obj:`None` the calculations are performed
| with respect to the center of the (changing) box.
| unwrap : bool
| When :obj:`True`, molecules that are broken due to the periodic boundary conditions
| are made whole.
|
| If the input contains molecules that are already whole, speed up the calculation by
| disabling unwrap. To do so, use the flag ``-no-unwrap`` when using MAICoS from the
| command line, or use ``unwrap=False`` when using MAICoS from the Python interpreter.
|
| Note: Molecules containing virtual sites (e.g. TIP4P water models) are not currently
| supported in MDAnalysis. In this case, you need to provide unwrapped trajectory
| files directly, and disable unwrap. Trajectories can be unwrapped, for example,
| using the ``trjconv`` command of GROMACS.
| pack : bool
| When :obj:`True`, molecules are put back into the unit cell. This is required
| because MAICoS only takes into account molecules that are inside the unit cell.
|
| If the input contains molecules that are already packed, speed up the calculation by
| disabling packing with ``pack=False``.
| jitter : float
| Magnitude of the random noise to add to the atomic positions.
|
| A jitter can be used to stabilize the aliasing effects sometimes appearing when
| histogramming data. The jitter value should be about the precision of the
| trajectory. In that case, using jitter will not alter the results of the histogram.
| If ``jitter = 0.0`` (default), the original atomic positions are kept unchanged.
|
| You can estimate the precision of the positions in your trajectory with
| :func:`maicos.lib.util.trajectory_precision`. Note that if the precision is not the
| same for all frames, the smallest precision should be used.
| concfreq : int
| When concfreq (for conclude frequency) is larger than ``0``, the conclude function
| is called and the output files are written every ``concfreq`` frames.
| output : str
| Output filename.
|
| Attributes
| ----------
| results.bin_pos : numpy.ndarray
| Bin positions (in Å) ranging from ``rmin`` to ``rmax``.
| results.profile : numpy.ndarray
| Calculated profile.
| results.dprofile : numpy.ndarray
| Estimated profile's uncertainity.
|
| Method resolution order:
| DiporderCylinder
| maicos.core.cylinder.ProfileCylinderBase
| maicos.core.cylinder.CylinderBase
| maicos.core.planar.PlanarBase
| maicos.core.base.AnalysisBase
| maicos.core.base._Runner
| MDAnalysis.analysis.base.AnalysisBase
| maicos.core.base.ProfileBase
| builtins.object
|
| Methods defined here:
|
| __init__(
| self,
| atomgroup: mda.AtomGroup,
| order_parameter: str = 'P1',
| pdim: str = 'r',
| dim: int = 2,
| zmin: float | None = None,
| zmax: float | None = None,
| rmin: float = 0,
| rmax: float | None = None,
| bin_width: float = 1,
| bin_method: str = 'com',
| grouping: str = 'residues',
| refgroup: mda.AtomGroup | None = None,
| unwrap: bool = True,
| pack: bool = True,
| jitter: float = 0.0,
| concfreq: int = 0,
| output: str = 'diporder_cylinder.dat'
| ) -> None
| Initialize self. See help(type(self)) for accurate signature.
|
| ----------------------------------------------------------------------
| Readonly properties inherited from maicos.core.planar.PlanarBase:
|
| odims
| Other dimensions perpendicular to dim i.e. (0,2) if dim = 1.
|
| ----------------------------------------------------------------------
| Methods inherited from maicos.core.base.AnalysisBase:
|
| dump(self, filename: str) -> None
| Save analysis state to an ``.npz`` file.
|
| .. warning::
|
| :meth:`dump` is **not** an archival format. The on-disk layout
| tracks the installed MAICoS and MDAnalysis versions and may stop
| loading after an upgrade. Use it to checkpoint or hand off an
| in-progress analysis, not to preserve results long-term. For
| archival output write summary tables with :meth:`save` or export
| :attr:`results` to a stable format.
|
| Persists all statistical accumulators (``means``, ``sems``, ``sums``,
| ``pop``, ``M2``), the ``results`` and ``_obs`` containers, per-frame
| arrays (``timeseries``, ``frames``, ``times``), metadata, the associated
| Universe and the analysed atomgroup. Restore the analysis with :meth:`load`.
|
| Parameters
| ----------
| filename : str
| Path to the output ``.npz`` file.
|
| run(
| self,
| start: int | None = None,
| stop: int | None = None,
| step: int | None = None,
| frames: int | None = None,
| verbose: bool | None = None,
| progressbar_kwargs: dict | None = None
| ) -> Self
| Iterate over the trajectory.
|
| Parameters
| ----------
| start : int
| start frame of analysis
| stop : int
| stop frame of analysis
| step : int
| number of frames to skip between each analysed frame
| frames : array_like
| array of integers or booleans to slice trajectory; ``frames`` can only be
| used *instead* of ``start``, ``stop``, and ``step``. Setting *both*
| ``frames`` and at least one of ``start``, ``stop``, ``step`` to a
| non-default value will raise a :exc:`ValueError`.
| verbose : bool
| Turn on verbosity
| progressbar_kwargs : dict
| ProgressBar keywords with custom parameters regarding progress bar position,
| etc; see :class:`MDAnalysis.lib.log.ProgressBar` for full list.
|
| Returns
| -------
| self : object
| analysis object
|
| savetxt(self, fname: str, X: np.ndarray, columns: list[str] | None = None) -> None
| Save to text.
|
| An extension of the numpy savetxt function. Adds the command line input to the
| header and checks for a doubled defined filesuffix.
|
| Return a header for the text file to save the data to. This method builds a
| generic header that can be used by any MAICoS module. It is called by the save
| method of each module.
|
| The information it collects is:
| - timestamp of the analysis
| - name of the module
| - version of MAICoS that was used
| - command line arguments that were used to run the module
| - module call including the default arguments
| - number of frames that were analyzed
| - atomgroup that was analyzed
| - output messages from modules and base classes (if they exist)
|
| ----------------------------------------------------------------------
| Class methods inherited from maicos.core.base.AnalysisBase:
|
| load(filename: str) -> Self
| Restore an analysis instance from a file created by :meth:`dump`.
|
| Returns a new instance of ``cls`` with all statistical accumulators,
| per-frame arrays, metadata, and the analysed atomgroup populated from
| the file. The rebuilt :class:`MDAnalysis.Universe` carries only the
| topology — no trajectory — so calling :meth:`run` on the returned
| instance raises :class:`RuntimeError`. Use the loaded instance to
| inspect :attr:`results` or to call :meth:`save`.
|
| Parameters
| ----------
| filename : str
| Path to the ``.npz`` file written by :meth:`dump`.
|
| Returns
| -------
| Self
| A new instance of ``cls`` with state restored from ``filename``.
|
| ----------------------------------------------------------------------
| Readonly properties inherited from maicos.core.base.AnalysisBase:
|
| box_center
| Center of the simulation cell.
|
| box_lengths
| Lengths of the simulation cell vectors.
|
| ----------------------------------------------------------------------
| Data descriptors inherited from maicos.core.base._Runner:
|
| __dict__
| dictionary for instance variables
|
| __weakref__
| list of weak references to the object
|
| ----------------------------------------------------------------------
| Class methods inherited from MDAnalysis.analysis.base.AnalysisBase:
|
| get_supported_backends()
| Tuple with backends supported by the core library for a given class.
| User can pass either one of these values as ``backend=...`` to
| :meth:`run()` method, or a custom object that has ``apply`` method
| (see documentation for :meth:`run()`):
|
| - 'serial': no parallelization
| - 'multiprocessing': parallelization using `multiprocessing.Pool`
| - 'dask': parallelization using `dask.delayed.compute()`. Requires
| installation of `mdanalysis[dask]`
|
| If you want to add your own backend to an existing class, pass a
| :class:`backends.BackendBase` subclass (see its documentation to learn
| how to implement it properly), and specify ``unsupported_backend=True``.
|
| Returns
| -------
| tuple
| names of built-in backends that can be used in :meth:`run(backend=...)`
|
|
| .. versionadded:: 2.8.0
|
| ----------------------------------------------------------------------
| Readonly properties inherited from MDAnalysis.analysis.base.AnalysisBase:
|
| parallelizable
| Boolean mark showing that a given class can be parallelizable with
| split-apply-combine procedure. Namely, if we can safely distribute
| :meth:`_single_frame` to multiple workers and then combine them with a
| proper :meth:`_conclude` call. If set to ``False``, no backends except
| for ``serial`` are supported.
|
| .. note:: If you want to check parallelizability of the whole class, without
| explicitly creating an instance of the class, see
| :attr:`_analysis_algorithm_is_parallelizable`. Note that you
| setting it to other value will break things if the algorithm
| behind the analysis is not trivially parallelizable.
|
|
| Returns
| -------
| bool
| if a given ``AnalysisBase`` subclass instance
| is parallelizable with split-apply-combine, or not
|
|
| .. versionadded:: 2.8.0
|
| ----------------------------------------------------------------------
| Methods inherited from maicos.core.base.ProfileBase:
|
| save(self) -> None
| Save results of analysis to file specified by ``output``.
class DiporderPlanar(maicos.core.planar.ProfilePlanarBase)
| DiporderPlanar(
| atomgroup: mda.AtomGroup,
| order_parameter: str = 'P1',
| pdim: int = 2,
| dim: int = 2,
| zmin: float | None = None,
| zmax: float | None = None,
| bin_width: float = 1,
| bin_method: str = 'com',
| grouping: str = 'residues',
| sym: bool = False,
| refgroup: mda.AtomGroup | None = None,
| unwrap: bool = True,
| pack: bool = True,
| jitter: float = 0.0,
| concfreq: int = 0,
| output: str = 'diporder_planar.dat'
| ) -> None
|
| Cartesian dipolar order parameters.
|
| Calculations include the first- and second-rank
| orientational order parameters,
| which are the Legendre polynomials :math:`P_1(x) = x` and :math:`P_2(x) = \frac{3
| x^2 - 1}{2}` with :math:`x = \cos(θ[z])`.
|
| For the correlation analysis the central bin
| (:math:`N / 2`) of the 0th's group profile is used. For further information on the correlation analysis please
| refer to :class:`AnalysisBase <maicos.core.base.AnalysisBase>` or the
| :ref:`general-design` section.
|
| Parameters
| ----------
| atomgroup : MDAnalysis.core.groups.AtomGroup
| A :class:`~MDAnalysis.core.groups.AtomGroup` for which the calculations are
| performed.
| order_parameter : {``"P1"``, ``"P2"``}
| Order parameter to be calculated:
| - ``"P1"``: Legendre polynomial :math:`P_1(cos[θ]) = cos[θ]`
| - ``"P2"``: Legendre polynomial
| :math:`P_2(cos[θ]) = \frac{3cos^2[θ] - 1}{2}`
| pdim : {0, 1, 2}
| direction of the projection
|
| dim : {0, 1, 2}
| Dimension for binning (``x=0``, ``y=1``, ``z=1``).
| zmin : float
| Minimal coordinate for evaluation (in Å) with respect to the center of mass of
| the refgroup.
| If ``zmin=None``, all coordinates down to the lower cell boundary are taken into
| account.
| zmax : float
| Maximal coordinate for evaluation (in Å) with respect to the center of mass of the
| refgroup.
| If ``zmax = None``, all coordinates up to the upper cell boundary are taken into
| account.
| bin_width : float
| Width of the bins (in Å).
| bin_method : {``"com"``, ``"cog"``, ``"coc"``}
| Method for the position binning.
|
| The possible options are center of mass (``"com"``), center of geometry (``"cog"``),
| and center of charge (``"coc"``).
| grouping : {``"atoms"``, ``"residues"``, ``"segments"``, ``"molecules"``, ``"fragments"``}
| Atom grouping for the calculations.
|
| The possible grouping options are the atom positions (in the case where
| ``grouping="atoms"``) or the center of mass of the specified grouping unit (in the
| case where ``grouping="residues"``, ``"segments"``, ``"molecules"`` or
| ``"fragments"``).
| sym : bool
| Symmetrize the profile. Only works in combination with ``refgroup``.
| refgroup : MDAnalysis.core.groups.AtomGroup
| Reference :class:`~MDAnalysis.core.groups.AtomGroup` used for the calculation. If
| ``refgroup`` is provided, the calculation is performed relative to the center of
| mass of the AtomGroup. If ``refgroup`` is :obj:`None` the calculations are performed
| with respect to the center of the (changing) box.
| unwrap : bool
| When :obj:`True`, molecules that are broken due to the periodic boundary conditions
| are made whole.
|
| If the input contains molecules that are already whole, speed up the calculation by
| disabling unwrap. To do so, use the flag ``-no-unwrap`` when using MAICoS from the
| command line, or use ``unwrap=False`` when using MAICoS from the Python interpreter.
|
| Note: Molecules containing virtual sites (e.g. TIP4P water models) are not currently
| supported in MDAnalysis. In this case, you need to provide unwrapped trajectory
| files directly, and disable unwrap. Trajectories can be unwrapped, for example,
| using the ``trjconv`` command of GROMACS.
| pack : bool
| When :obj:`True`, molecules are put back into the unit cell. This is required
| because MAICoS only takes into account molecules that are inside the unit cell.
|
| If the input contains molecules that are already packed, speed up the calculation by
| disabling packing with ``pack=False``.
| jitter : float
| Magnitude of the random noise to add to the atomic positions.
|
| A jitter can be used to stabilize the aliasing effects sometimes appearing when
| histogramming data. The jitter value should be about the precision of the
| trajectory. In that case, using jitter will not alter the results of the histogram.
| If ``jitter = 0.0`` (default), the original atomic positions are kept unchanged.
|
| You can estimate the precision of the positions in your trajectory with
| :func:`maicos.lib.util.trajectory_precision`. Note that if the precision is not the
| same for all frames, the smallest precision should be used.
| concfreq : int
| When concfreq (for conclude frequency) is larger than ``0``, the conclude function
| is called and the output files are written every ``concfreq`` frames.
| output : str
| Output filename.
|
| Attributes
| ----------
| results.bin_pos : numpy.ndarray
| Bin positions (in Å) ranging from ``zmin`` to ``zmax``.
| results.profile : numpy.ndarray
| Calculated profile.
| results.dprofile : numpy.ndarray
| Estimated profile's uncertainity.
|
| Method resolution order:
| DiporderPlanar
| maicos.core.planar.ProfilePlanarBase
| maicos.core.planar.PlanarBase
| maicos.core.base.AnalysisBase
| maicos.core.base._Runner
| MDAnalysis.analysis.base.AnalysisBase
| maicos.core.base.ProfileBase
| builtins.object
|
| Methods defined here:
|
| __init__(
| self,
| atomgroup: mda.AtomGroup,
| order_parameter: str = 'P1',
| pdim: int = 2,
| dim: int = 2,
| zmin: float | None = None,
| zmax: float | None = None,
| bin_width: float = 1,
| bin_method: str = 'com',
| grouping: str = 'residues',
| sym: bool = False,
| refgroup: mda.AtomGroup | None = None,
| unwrap: bool = True,
| pack: bool = True,
| jitter: float = 0.0,
| concfreq: int = 0,
| output: str = 'diporder_planar.dat'
| ) -> None
| Initialize self. See help(type(self)) for accurate signature.
|
| ----------------------------------------------------------------------
| Readonly properties inherited from maicos.core.planar.PlanarBase:
|
| odims
| Other dimensions perpendicular to dim i.e. (0,2) if dim = 1.
|
| ----------------------------------------------------------------------
| Methods inherited from maicos.core.base.AnalysisBase:
|
| dump(self, filename: str) -> None
| Save analysis state to an ``.npz`` file.
|
| .. warning::
|
| :meth:`dump` is **not** an archival format. The on-disk layout
| tracks the installed MAICoS and MDAnalysis versions and may stop
| loading after an upgrade. Use it to checkpoint or hand off an
| in-progress analysis, not to preserve results long-term. For
| archival output write summary tables with :meth:`save` or export
| :attr:`results` to a stable format.
|
| Persists all statistical accumulators (``means``, ``sems``, ``sums``,
| ``pop``, ``M2``), the ``results`` and ``_obs`` containers, per-frame
| arrays (``timeseries``, ``frames``, ``times``), metadata, the associated
| Universe and the analysed atomgroup. Restore the analysis with :meth:`load`.
|
| Parameters
| ----------
| filename : str
| Path to the output ``.npz`` file.
|
| run(
| self,
| start: int | None = None,
| stop: int | None = None,
| step: int | None = None,
| frames: int | None = None,
| verbose: bool | None = None,
| progressbar_kwargs: dict | None = None
| ) -> Self
| Iterate over the trajectory.
|
| Parameters
| ----------
| start : int
| start frame of analysis
| stop : int
| stop frame of analysis
| step : int
| number of frames to skip between each analysed frame
| frames : array_like
| array of integers or booleans to slice trajectory; ``frames`` can only be
| used *instead* of ``start``, ``stop``, and ``step``. Setting *both*
| ``frames`` and at least one of ``start``, ``stop``, ``step`` to a
| non-default value will raise a :exc:`ValueError`.
| verbose : bool
| Turn on verbosity
| progressbar_kwargs : dict
| ProgressBar keywords with custom parameters regarding progress bar position,
| etc; see :class:`MDAnalysis.lib.log.ProgressBar` for full list.
|
| Returns
| -------
| self : object
| analysis object
|
| savetxt(self, fname: str, X: np.ndarray, columns: list[str] | None = None) -> None
| Save to text.
|
| An extension of the numpy savetxt function. Adds the command line input to the
| header and checks for a doubled defined filesuffix.
|
| Return a header for the text file to save the data to. This method builds a
| generic header that can be used by any MAICoS module. It is called by the save
| method of each module.
|
| The information it collects is:
| - timestamp of the analysis
| - name of the module
| - version of MAICoS that was used
| - command line arguments that were used to run the module
| - module call including the default arguments
| - number of frames that were analyzed
| - atomgroup that was analyzed
| - output messages from modules and base classes (if they exist)
|
| ----------------------------------------------------------------------
| Class methods inherited from maicos.core.base.AnalysisBase:
|
| load(filename: str) -> Self
| Restore an analysis instance from a file created by :meth:`dump`.
|
| Returns a new instance of ``cls`` with all statistical accumulators,
| per-frame arrays, metadata, and the analysed atomgroup populated from
| the file. The rebuilt :class:`MDAnalysis.Universe` carries only the
| topology — no trajectory — so calling :meth:`run` on the returned
| instance raises :class:`RuntimeError`. Use the loaded instance to
| inspect :attr:`results` or to call :meth:`save`.
|
| Parameters
| ----------
| filename : str
| Path to the ``.npz`` file written by :meth:`dump`.
|
| Returns
| -------
| Self
| A new instance of ``cls`` with state restored from ``filename``.
|
| ----------------------------------------------------------------------
| Readonly properties inherited from maicos.core.base.AnalysisBase:
|
| box_center
| Center of the simulation cell.
|
| box_lengths
| Lengths of the simulation cell vectors.
|
| ----------------------------------------------------------------------
| Data descriptors inherited from maicos.core.base._Runner:
|
| __dict__
| dictionary for instance variables
|
| __weakref__
| list of weak references to the object
|
| ----------------------------------------------------------------------
| Class methods inherited from MDAnalysis.analysis.base.AnalysisBase:
|
| get_supported_backends()
| Tuple with backends supported by the core library for a given class.
| User can pass either one of these values as ``backend=...`` to
| :meth:`run()` method, or a custom object that has ``apply`` method
| (see documentation for :meth:`run()`):
|
| - 'serial': no parallelization
| - 'multiprocessing': parallelization using `multiprocessing.Pool`
| - 'dask': parallelization using `dask.delayed.compute()`. Requires
| installation of `mdanalysis[dask]`
|
| If you want to add your own backend to an existing class, pass a
| :class:`backends.BackendBase` subclass (see its documentation to learn
| how to implement it properly), and specify ``unsupported_backend=True``.
|
| Returns
| -------
| tuple
| names of built-in backends that can be used in :meth:`run(backend=...)`
|
|
| .. versionadded:: 2.8.0
|
| ----------------------------------------------------------------------
| Readonly properties inherited from MDAnalysis.analysis.base.AnalysisBase:
|
| parallelizable
| Boolean mark showing that a given class can be parallelizable with
| split-apply-combine procedure. Namely, if we can safely distribute
| :meth:`_single_frame` to multiple workers and then combine them with a
| proper :meth:`_conclude` call. If set to ``False``, no backends except
| for ``serial`` are supported.
|
| .. note:: If you want to check parallelizability of the whole class, without
| explicitly creating an instance of the class, see
| :attr:`_analysis_algorithm_is_parallelizable`. Note that you
| setting it to other value will break things if the algorithm
| behind the analysis is not trivially parallelizable.
|
|
| Returns
| -------
| bool
| if a given ``AnalysisBase`` subclass instance
| is parallelizable with split-apply-combine, or not
|
|
| .. versionadded:: 2.8.0
|
| ----------------------------------------------------------------------
| Methods inherited from maicos.core.base.ProfileBase:
|
| save(self) -> None
| Save results of analysis to file specified by ``output``.
class DiporderSphere(maicos.core.sphere.ProfileSphereBase)
| DiporderSphere(
| atomgroup: mda.AtomGroup,
| order_parameter: str = 'P1',
| rmin: float = 0,
| rmax: float | None = None,
| bin_width: float = 1,
| bin_method: str = 'com',
| grouping: str = 'residues',
| refgroup: mda.AtomGroup | None = None,
| unwrap: bool = True,
| pack: bool = True,
| jitter: float = 0.0,
| concfreq: int = 0,
| output: str = 'diporder_sphere.dat'
| ) -> None
|
| Spherical dipolar order parameters.
|
| Calculations include the first- and second-rank
| orientational order parameters,
| which are the Legendre polynomials :math:`P_1(x) = x` and :math:`P_2(x) = \frac{3
| x^2 - 1}{2}` with :math:`x = \cos(θ[z])`.
|
| For the correlation analysis the 0th bin of the 0th's
| group profile is used. For further information on the correlation analysis please
| refer to :class:`AnalysisBase <maicos.core.base.AnalysisBase>` or the
| :ref:`general-design` section.
|
| Parameters
| ----------
| atomgroup : MDAnalysis.core.groups.AtomGroup
| A :class:`~MDAnalysis.core.groups.AtomGroup` for which the calculations are
| performed.
| order_parameter : {``"P1"``, ``"P2"``}
| Order parameter to be calculated:
| - ``"P1"``: Legendre polynomial :math:`P_1(cos[θ]) = cos[θ]`
| - ``"P2"``: Legendre polynomial
| :math:`P_2(cos[θ]) = \frac{3cos^2[θ] - 1}{2}`
| rmin : float
| Minimal radial coordinate relative to the center of mass of the refgroup for
| evaluation (in Å).
| rmax : float
| Maximal radial coordinate relative to the center of mass of the refgroup for
| evaluation (in Å).
|
| If ``rmax=None``, the box extension is taken.
| bin_width : float
| Width of the bins (in Å).
| bin_method : {``"com"``, ``"cog"``, ``"coc"``}
| Method for the position binning.
|
| The possible options are center of mass (``"com"``), center of geometry (``"cog"``),
| and center of charge (``"coc"``).
| grouping : {``"atoms"``, ``"residues"``, ``"segments"``, ``"molecules"``, ``"fragments"``}
| Atom grouping for the calculations.
|
| The possible grouping options are the atom positions (in the case where
| ``grouping="atoms"``) or the center of mass of the specified grouping unit (in the
| case where ``grouping="residues"``, ``"segments"``, ``"molecules"`` or
| ``"fragments"``).
| refgroup : MDAnalysis.core.groups.AtomGroup
| Reference :class:`~MDAnalysis.core.groups.AtomGroup` used for the calculation. If
| ``refgroup`` is provided, the calculation is performed relative to the center of
| mass of the AtomGroup. If ``refgroup`` is :obj:`None` the calculations are performed
| with respect to the center of the (changing) box.
| unwrap : bool
| When :obj:`True`, molecules that are broken due to the periodic boundary conditions
| are made whole.
|
| If the input contains molecules that are already whole, speed up the calculation by
| disabling unwrap. To do so, use the flag ``-no-unwrap`` when using MAICoS from the
| command line, or use ``unwrap=False`` when using MAICoS from the Python interpreter.
|
| Note: Molecules containing virtual sites (e.g. TIP4P water models) are not currently
| supported in MDAnalysis. In this case, you need to provide unwrapped trajectory
| files directly, and disable unwrap. Trajectories can be unwrapped, for example,
| using the ``trjconv`` command of GROMACS.
| pack : bool
| When :obj:`True`, molecules are put back into the unit cell. This is required
| because MAICoS only takes into account molecules that are inside the unit cell.
|
| If the input contains molecules that are already packed, speed up the calculation by
| disabling packing with ``pack=False``.
| jitter : float
| Magnitude of the random noise to add to the atomic positions.
|
| A jitter can be used to stabilize the aliasing effects sometimes appearing when
| histogramming data. The jitter value should be about the precision of the
| trajectory. In that case, using jitter will not alter the results of the histogram.
| If ``jitter = 0.0`` (default), the original atomic positions are kept unchanged.
|
| You can estimate the precision of the positions in your trajectory with
| :func:`maicos.lib.util.trajectory_precision`. Note that if the precision is not the
| same for all frames, the smallest precision should be used.
| concfreq : int
| When concfreq (for conclude frequency) is larger than ``0``, the conclude function
| is called and the output files are written every ``concfreq`` frames.
| output : str
| Output filename.
|
| Attributes
| ----------
| results.bin_pos : numpy.ndarray
| Bin positions (in Å) ranging from ``rmin`` to ``rmax``.
| results.profile : numpy.ndarray
| Calculated profile.
| results.dprofile : numpy.ndarray
| Estimated profile's uncertainity.
|
| Method resolution order:
| DiporderSphere
| maicos.core.sphere.ProfileSphereBase
| maicos.core.sphere.SphereBase
| maicos.core.base.AnalysisBase
| maicos.core.base._Runner
| MDAnalysis.analysis.base.AnalysisBase
| maicos.core.base.ProfileBase
| builtins.object
|
| Methods defined here:
|
| __init__(
| self,
| atomgroup: mda.AtomGroup,
| order_parameter: str = 'P1',
| rmin: float = 0,
| rmax: float | None = None,
| bin_width: float = 1,
| bin_method: str = 'com',
| grouping: str = 'residues',
| refgroup: mda.AtomGroup | None = None,
| unwrap: bool = True,
| pack: bool = True,
| jitter: float = 0.0,
| concfreq: int = 0,
| output: str = 'diporder_sphere.dat'
| ) -> None
| Initialize self. See help(type(self)) for accurate signature.
|
| ----------------------------------------------------------------------
| Methods inherited from maicos.core.base.AnalysisBase:
|
| dump(self, filename: str) -> None
| Save analysis state to an ``.npz`` file.
|
| .. warning::
|
| :meth:`dump` is **not** an archival format. The on-disk layout
| tracks the installed MAICoS and MDAnalysis versions and may stop
| loading after an upgrade. Use it to checkpoint or hand off an
| in-progress analysis, not to preserve results long-term. For
| archival output write summary tables with :meth:`save` or export
| :attr:`results` to a stable format.
|
| Persists all statistical accumulators (``means``, ``sems``, ``sums``,
| ``pop``, ``M2``), the ``results`` and ``_obs`` containers, per-frame
| arrays (``timeseries``, ``frames``, ``times``), metadata, the associated
| Universe and the analysed atomgroup. Restore the analysis with :meth:`load`.
|
| Parameters
| ----------
| filename : str
| Path to the output ``.npz`` file.
|
| run(
| self,
| start: int | None = None,
| stop: int | None = None,
| step: int | None = None,
| frames: int | None = None,
| verbose: bool | None = None,
| progressbar_kwargs: dict | None = None
| ) -> Self
| Iterate over the trajectory.
|
| Parameters
| ----------
| start : int
| start frame of analysis
| stop : int
| stop frame of analysis
| step : int
| number of frames to skip between each analysed frame
| frames : array_like
| array of integers or booleans to slice trajectory; ``frames`` can only be
| used *instead* of ``start``, ``stop``, and ``step``. Setting *both*
| ``frames`` and at least one of ``start``, ``stop``, ``step`` to a
| non-default value will raise a :exc:`ValueError`.
| verbose : bool
| Turn on verbosity
| progressbar_kwargs : dict
| ProgressBar keywords with custom parameters regarding progress bar position,
| etc; see :class:`MDAnalysis.lib.log.ProgressBar` for full list.
|
| Returns
| -------
| self : object
| analysis object
|
| savetxt(self, fname: str, X: np.ndarray, columns: list[str] | None = None) -> None
| Save to text.
|
| An extension of the numpy savetxt function. Adds the command line input to the
| header and checks for a doubled defined filesuffix.
|
| Return a header for the text file to save the data to. This method builds a
| generic header that can be used by any MAICoS module. It is called by the save
| method of each module.
|
| The information it collects is:
| - timestamp of the analysis
| - name of the module
| - version of MAICoS that was used
| - command line arguments that were used to run the module
| - module call including the default arguments
| - number of frames that were analyzed
| - atomgroup that was analyzed
| - output messages from modules and base classes (if they exist)
|
| ----------------------------------------------------------------------
| Class methods inherited from maicos.core.base.AnalysisBase:
|
| load(filename: str) -> Self
| Restore an analysis instance from a file created by :meth:`dump`.
|
| Returns a new instance of ``cls`` with all statistical accumulators,
| per-frame arrays, metadata, and the analysed atomgroup populated from
| the file. The rebuilt :class:`MDAnalysis.Universe` carries only the
| topology — no trajectory — so calling :meth:`run` on the returned
| instance raises :class:`RuntimeError`. Use the loaded instance to
| inspect :attr:`results` or to call :meth:`save`.
|
| Parameters
| ----------
| filename : str
| Path to the ``.npz`` file written by :meth:`dump`.
|
| Returns
| -------
| Self
| A new instance of ``cls`` with state restored from ``filename``.
|
| ----------------------------------------------------------------------
| Readonly properties inherited from maicos.core.base.AnalysisBase:
|
| box_center
| Center of the simulation cell.
|
| box_lengths
| Lengths of the simulation cell vectors.
|
| ----------------------------------------------------------------------
| Data descriptors inherited from maicos.core.base._Runner:
|
| __dict__
| dictionary for instance variables
|
| __weakref__
| list of weak references to the object
|
| ----------------------------------------------------------------------
| Class methods inherited from MDAnalysis.analysis.base.AnalysisBase:
|
| get_supported_backends()
| Tuple with backends supported by the core library for a given class.
| User can pass either one of these values as ``backend=...`` to
| :meth:`run()` method, or a custom object that has ``apply`` method
| (see documentation for :meth:`run()`):
|
| - 'serial': no parallelization
| - 'multiprocessing': parallelization using `multiprocessing.Pool`
| - 'dask': parallelization using `dask.delayed.compute()`. Requires
| installation of `mdanalysis[dask]`
|
| If you want to add your own backend to an existing class, pass a
| :class:`backends.BackendBase` subclass (see its documentation to learn
| how to implement it properly), and specify ``unsupported_backend=True``.
|
| Returns
| -------
| tuple
| names of built-in backends that can be used in :meth:`run(backend=...)`
|
|
| .. versionadded:: 2.8.0
|
| ----------------------------------------------------------------------
| Readonly properties inherited from MDAnalysis.analysis.base.AnalysisBase:
|
| parallelizable
| Boolean mark showing that a given class can be parallelizable with
| split-apply-combine procedure. Namely, if we can safely distribute
| :meth:`_single_frame` to multiple workers and then combine them with a
| proper :meth:`_conclude` call. If set to ``False``, no backends except
| for ``serial`` are supported.
|
| .. note:: If you want to check parallelizability of the whole class, without
| explicitly creating an instance of the class, see
| :attr:`_analysis_algorithm_is_parallelizable`. Note that you
| setting it to other value will break things if the algorithm
| behind the analysis is not trivially parallelizable.
|
|
| Returns
| -------
| bool
| if a given ``AnalysisBase`` subclass instance
| is parallelizable with split-apply-combine, or not
|
|
| .. versionadded:: 2.8.0
|
| ----------------------------------------------------------------------
| Methods inherited from maicos.core.base.ProfileBase:
|
| save(self) -> None
| Save results of analysis to file specified by ``output``.
class DiporderStructureFactor(builtins.object)
| DiporderStructureFactor(
| atomgroup: mda.AtomGroup,
| qmin: float = 0,
| qmax: float = 6,
| dq: float = 0.01,
| bin_method: str = 'com',
| grouping: str = 'molecules',
| refgroup: mda.AtomGroup | None = None,
| unwrap: bool = True,
| pack: bool = True,
| jitter: float = 0.0,
| concfreq: int = 0,
| output: str = 'sq.dat'
| ) -> None
|
| Class has been moved to the ``scatterkit`` package.
|
| Methods defined here:
|
| __init__(
| self,
| atomgroup: mda.AtomGroup,
| qmin: float = 0,
| qmax: float = 6,
| dq: float = 0.01,
| bin_method: str = 'com',
| grouping: str = 'molecules',
| refgroup: mda.AtomGroup | None = None,
| unwrap: bool = True,
| pack: bool = True,
| jitter: float = 0.0,
| concfreq: int = 0,
| output: str = 'sq.dat'
| ) -> None
| Initialize self. See help(type(self)) for accurate signature.
|
| ----------------------------------------------------------------------
| Data descriptors defined here:
|
| __dict__
| dictionary for instance variables
|
| __weakref__
| list of weak references to the object
class KineticEnergy(builtins.object)
| KineticEnergy(
| atomgroup: mda.AtomGroup,
| refpoint: str = 'com',
| refgroup: mda.AtomGroup | None = None,
| unwrap: bool = False,
| pack: bool = True,
| jitter: float = 0.0,
| concfreq: int = 0,
| output: str = 'ke.dat'
| ) -> None
|
| Class has been removed and is no longer available.
|
| Methods defined here:
|
| __init__(
| self,
| atomgroup: mda.AtomGroup,
| refpoint: str = 'com',
| refgroup: mda.AtomGroup | None = None,
| unwrap: bool = False,
| pack: bool = True,
| jitter: float = 0.0,
| concfreq: int = 0,
| output: str = 'ke.dat'
| ) -> None
| Initialize self. See help(type(self)) for accurate signature.
|
| ----------------------------------------------------------------------
| Data descriptors defined here:
|
| __dict__
| dictionary for instance variables
|
| __weakref__
| list of weak references to the object
class PDFCylinder(maicos.core.cylinder.CylinderBase)
| PDFCylinder(
| g1: mda.AtomGroup,
| g2: mda.AtomGroup | None = None,
| bin_width_pdf_z: float = 0.3,
| bin_width_pdf_phi: float = 0.1,
| drwidth: float = 0.1,
| dmin: float | None = None,
| dmax: float | None = None,
| density: bool = False,
| origin: np.ndarray | None = None,
| dim: int = 2,
| zmin: float | None = None,
| zmax: float | None = None,
| rmin: float = 0,
| rmax: float | None = None,
| bin_width: float = 1,
| bin_method: str = 'com',
| refgroup: mda.AtomGroup | None = None,
| unwrap: bool = False,
| pack: bool = True,
| jitter: float = 0.0,
| concfreq: int = 0,
| output: str = 'pdf.dat'
| ) -> None
|
| Shell-wise one-dimensional (cylindrical) pair distribution functions.
|
| The one-dimensional pair distribution functions :math:`g_{\text{1d}}(\phi)`
| and :math:`g_{\text{1d}}(z)` describes the pair distribution to particles
| which lie on the same cylinder along the angular and axial directions
| respectively. These functions can be used in cylindrical systems that are
| inhomogeneous along radial coordinate, and homogeneous in the angular and
| axial directions. It gives the average number density of ``g2`` as a
| function of angular and axial distances respectively from a ``g1`` atom.
| Then the angular pair distribution function is
|
| .. math::
|
| g_{\text{1d}}(\phi) = \left \langle \sum_{i}^{N_{g_1}}
| \sum_{j}^{N_{g2}} \delta(\phi - \phi_{ij}) \delta(R_{ij}) \delta(z_{ij})
| \right \rangle
|
|
| And the axial pair distribution function is
|
| .. math::
|
| g_{\text{1d}}(z) = \left \langle \sum_{i}^{N_{g_1}}
| \sum_{j}^{N_{g2}} \delta(z - z_{ij}) \delta(R_{ij}) \delta(\phi_{ij})
| \right \rangle
|
| Even though due to consistency reasons the results are called pair distribution
| functions the output is not unitless. The default output is is in dimension of
| number/volume in :math:`Å^{-3}`. If ``density`` is set to :py:obj:`True`, the
| output is normalised by the density of ``g2``.
|
| Parameters
| ----------
| atomgroup : MDAnalysis.core.groups.AtomGroup
| A :class:`~MDAnalysis.core.groups.AtomGroup` for which the calculations are
| performed.
| g1 : MDAnalysis.core.groups.AtomGroup
| First AtomGroup.
| g2 : MDAnalysis.core.groups.AtomGroup
| Second AtomGroup.
| bin_width_pdf_z : float
| Binwidth of bins in the histogram of the axial PDF (Å).
| bin_width_pdf_phi : float
| Binwidth of bins in the histogram of the angular PDF (Å).
| drwidth : float
| radial width of a PDF cylindrical shell (Å), and axial or angular (arc) slices.
| dmin: float
| the minimum pairwise distance between ``'g1'`` and ``'g2'`` (Å).
| dmax : float
| the maximum pairwise distance between ``'g1'`` and ``'g2'`` (Å).
| density : bool
| normalise the PDF by the density of ``'g2'`` (:math:`Å^{-3}`).
| origin : numpy.ndarray
| Set origin of the cylindrical coordinate system (x,y,z). If :obj:`None` the
| origin will be set according to the ``refgroup`` parameter.
| dim : {0, 1, 2}
| Dimension for binning (``x=0``, ``y=1``, ``z=1``).
| zmin : float
| Minimal coordinate for evaluation (in Å) with respect to the center of mass of
| the refgroup.
| If ``zmin=None``, all coordinates down to the lower cell boundary are taken into
| account.
| zmax : float
| Maximal coordinate for evaluation (in Å) with respect to the center of mass of the
| refgroup.
| If ``zmax = None``, all coordinates up to the upper cell boundary are taken into
| account.
| rmin : float
| Minimal radial coordinate relative to the center of mass of the refgroup for
| evaluation (in Å).
| rmax : float
| Maximal radial coordinate relative to the center of mass of the refgroup for
| evaluation (in Å).
|
| If ``rmax=None``, the box extension is taken.
| bin_width : float
| Width of the bins (in Å).
| bin_method : {``"com"``, ``"cog"``, ``"coc"``}
| Method for the position binning.
|
| The possible options are center of mass (``"com"``), center of geometry (``"cog"``),
| and center of charge (``"coc"``).
| refgroup : MDAnalysis.core.groups.AtomGroup
| Reference :class:`~MDAnalysis.core.groups.AtomGroup` used for the calculation. If
| ``refgroup`` is provided, the calculation is performed relative to the center of
| mass of the AtomGroup. If ``refgroup`` is :obj:`None` the calculations are performed
| with respect to the center of the (changing) box.
| unwrap : bool
| When :obj:`True`, molecules that are broken due to the periodic boundary conditions
| are made whole.
|
| If the input contains molecules that are already whole, speed up the calculation by
| disabling unwrap. To do so, use the flag ``-no-unwrap`` when using MAICoS from the
| command line, or use ``unwrap=False`` when using MAICoS from the Python interpreter.
|
| Note: Molecules containing virtual sites (e.g. TIP4P water models) are not currently
| supported in MDAnalysis. In this case, you need to provide unwrapped trajectory
| files directly, and disable unwrap. Trajectories can be unwrapped, for example,
| using the ``trjconv`` command of GROMACS.
| pack : bool
| When :obj:`True`, molecules are put back into the unit cell. This is required
| because MAICoS only takes into account molecules that are inside the unit cell.
|
| If the input contains molecules that are already packed, speed up the calculation by
| disabling packing with ``pack=False``.
| jitter : float
| Magnitude of the random noise to add to the atomic positions.
|
| A jitter can be used to stabilize the aliasing effects sometimes appearing when
| histogramming data. The jitter value should be about the precision of the
| trajectory. In that case, using jitter will not alter the results of the histogram.
| If ``jitter = 0.0`` (default), the original atomic positions are kept unchanged.
|
| You can estimate the precision of the positions in your trajectory with
| :func:`maicos.lib.util.trajectory_precision`. Note that if the precision is not the
| same for all frames, the smallest precision should be used.
| concfreq : int
| When concfreq (for conclude frequency) is larger than ``0``, the conclude function
| is called and the output files are written every ``concfreq`` frames.
| output : str
| Output filename.
|
| Attributes
| ----------
| results.bin_pos : numpy.ndarray
| Bin positions (in Å) ranging from ``rmin`` to ``rmax``.
| results.phi_bins: numpy.ndarray
| Angular distances to which the PDF is calculated with shape (``pdf_nbins``) (Å)
| results.z_bins: numpy.ndarray
| axial distances to which the PDF is calculated with shape (``pdf_nbins``) (Å)
| results.phi_pdf: numpy.ndarray
| Angular PDF with shape (``pdf_nbins``, ``n_bins``) (:math:`\text{Å}^{-3}`)
| results.z_pdf: numpy.ndarray
| Axial PDF with shape (``pdf_nbins``, ``n_bins``) (:math:`\text{Å}^{-3}`)
|
| Method resolution order:
| PDFCylinder
| maicos.core.cylinder.CylinderBase
| maicos.core.planar.PlanarBase
| maicos.core.base.AnalysisBase
| maicos.core.base._Runner
| MDAnalysis.analysis.base.AnalysisBase
| builtins.object
|
| Methods defined here:
|
| __init__(
| self,
| g1: mda.AtomGroup,
| g2: mda.AtomGroup | None = None,
| bin_width_pdf_z: float = 0.3,
| bin_width_pdf_phi: float = 0.1,
| drwidth: float = 0.1,
| dmin: float | None = None,
| dmax: float | None = None,
| density: bool = False,
| origin: np.ndarray | None = None,
| dim: int = 2,
| zmin: float | None = None,
| zmax: float | None = None,
| rmin: float = 0,
| rmax: float | None = None,
| bin_width: float = 1,
| bin_method: str = 'com',
| refgroup: mda.AtomGroup | None = None,
| unwrap: bool = False,
| pack: bool = True,
| jitter: float = 0.0,
| concfreq: int = 0,
| output: str = 'pdf.dat'
| ) -> None
| Initialize self. See help(type(self)) for accurate signature.
|
| save(self) -> None
| Save results of analysis to file specified by ``output``.
|
| ----------------------------------------------------------------------
| Readonly properties inherited from maicos.core.planar.PlanarBase:
|
| odims
| Other dimensions perpendicular to dim i.e. (0,2) if dim = 1.
|
| ----------------------------------------------------------------------
| Methods inherited from maicos.core.base.AnalysisBase:
|
| dump(self, filename: str) -> None
| Save analysis state to an ``.npz`` file.
|
| .. warning::
|
| :meth:`dump` is **not** an archival format. The on-disk layout
| tracks the installed MAICoS and MDAnalysis versions and may stop
| loading after an upgrade. Use it to checkpoint or hand off an
| in-progress analysis, not to preserve results long-term. For
| archival output write summary tables with :meth:`save` or export
| :attr:`results` to a stable format.
|
| Persists all statistical accumulators (``means``, ``sems``, ``sums``,
| ``pop``, ``M2``), the ``results`` and ``_obs`` containers, per-frame
| arrays (``timeseries``, ``frames``, ``times``), metadata, the associated
| Universe and the analysed atomgroup. Restore the analysis with :meth:`load`.
|
| Parameters
| ----------
| filename : str
| Path to the output ``.npz`` file.
|
| run(
| self,
| start: int | None = None,
| stop: int | None = None,
| step: int | None = None,
| frames: int | None = None,
| verbose: bool | None = None,
| progressbar_kwargs: dict | None = None
| ) -> Self
| Iterate over the trajectory.
|
| Parameters
| ----------
| start : int
| start frame of analysis
| stop : int
| stop frame of analysis
| step : int
| number of frames to skip between each analysed frame
| frames : array_like
| array of integers or booleans to slice trajectory; ``frames`` can only be
| used *instead* of ``start``, ``stop``, and ``step``. Setting *both*
| ``frames`` and at least one of ``start``, ``stop``, ``step`` to a
| non-default value will raise a :exc:`ValueError`.
| verbose : bool
| Turn on verbosity
| progressbar_kwargs : dict
| ProgressBar keywords with custom parameters regarding progress bar position,
| etc; see :class:`MDAnalysis.lib.log.ProgressBar` for full list.
|
| Returns
| -------
| self : object
| analysis object
|
| savetxt(self, fname: str, X: np.ndarray, columns: list[str] | None = None) -> None
| Save to text.
|
| An extension of the numpy savetxt function. Adds the command line input to the
| header and checks for a doubled defined filesuffix.
|
| Return a header for the text file to save the data to. This method builds a
| generic header that can be used by any MAICoS module. It is called by the save
| method of each module.
|
| The information it collects is:
| - timestamp of the analysis
| - name of the module
| - version of MAICoS that was used
| - command line arguments that were used to run the module
| - module call including the default arguments
| - number of frames that were analyzed
| - atomgroup that was analyzed
| - output messages from modules and base classes (if they exist)
|
| ----------------------------------------------------------------------
| Class methods inherited from maicos.core.base.AnalysisBase:
|
| load(filename: str) -> Self
| Restore an analysis instance from a file created by :meth:`dump`.
|
| Returns a new instance of ``cls`` with all statistical accumulators,
| per-frame arrays, metadata, and the analysed atomgroup populated from
| the file. The rebuilt :class:`MDAnalysis.Universe` carries only the
| topology — no trajectory — so calling :meth:`run` on the returned
| instance raises :class:`RuntimeError`. Use the loaded instance to
| inspect :attr:`results` or to call :meth:`save`.
|
| Parameters
| ----------
| filename : str
| Path to the ``.npz`` file written by :meth:`dump`.
|
| Returns
| -------
| Self
| A new instance of ``cls`` with state restored from ``filename``.
|
| ----------------------------------------------------------------------
| Readonly properties inherited from maicos.core.base.AnalysisBase:
|
| box_center
| Center of the simulation cell.
|
| box_lengths
| Lengths of the simulation cell vectors.
|
| ----------------------------------------------------------------------
| Data descriptors inherited from maicos.core.base._Runner:
|
| __dict__
| dictionary for instance variables
|
| __weakref__
| list of weak references to the object
|
| ----------------------------------------------------------------------
| Class methods inherited from MDAnalysis.analysis.base.AnalysisBase:
|
| get_supported_backends()
| Tuple with backends supported by the core library for a given class.
| User can pass either one of these values as ``backend=...`` to
| :meth:`run()` method, or a custom object that has ``apply`` method
| (see documentation for :meth:`run()`):
|
| - 'serial': no parallelization
| - 'multiprocessing': parallelization using `multiprocessing.Pool`
| - 'dask': parallelization using `dask.delayed.compute()`. Requires
| installation of `mdanalysis[dask]`
|
| If you want to add your own backend to an existing class, pass a
| :class:`backends.BackendBase` subclass (see its documentation to learn
| how to implement it properly), and specify ``unsupported_backend=True``.
|
| Returns
| -------
| tuple
| names of built-in backends that can be used in :meth:`run(backend=...)`
|
|
| .. versionadded:: 2.8.0
|
| ----------------------------------------------------------------------
| Readonly properties inherited from MDAnalysis.analysis.base.AnalysisBase:
|
| parallelizable
| Boolean mark showing that a given class can be parallelizable with
| split-apply-combine procedure. Namely, if we can safely distribute
| :meth:`_single_frame` to multiple workers and then combine them with a
| proper :meth:`_conclude` call. If set to ``False``, no backends except
| for ``serial`` are supported.
|
| .. note:: If you want to check parallelizability of the whole class, without
| explicitly creating an instance of the class, see
| :attr:`_analysis_algorithm_is_parallelizable`. Note that you
| setting it to other value will break things if the algorithm
| behind the analysis is not trivially parallelizable.
|
|
| Returns
| -------
| bool
| if a given ``AnalysisBase`` subclass instance
| is parallelizable with split-apply-combine, or not
|
|
| .. versionadded:: 2.8.0
class PDFPlanar(maicos.core.planar.PlanarBase)
| PDFPlanar(
| g1: mda.AtomGroup,
| g2: mda.AtomGroup | None = None,
| pdf_bin_width: float = 0.3,
| dzheight: float = 0.1,
| dmin: float = 0.0,
| dmax: float | None = None,
| dim: int = 2,
| zmin: float | None = None,
| zmax: float | None = None,
| bin_width: float = 1,
| bin_method: str = 'com',
| refgroup: mda.AtomGroup | None = None,
| unwrap: bool = False,
| pack: bool = True,
| jitter: float = 0.0,
| concfreq: int = 0,
| output: str = 'pdf.dat'
| ) -> None
|
| Slab-wise planar 2D pair distribution functions.
|
| The pair distribution function :math:`g_\mathrm{2D}(r)` describes the
| spatial correlation between atoms in :math:`g_1` and atoms in
| :math:`g_2`, which lie in the same plane.
| It gives the average number density of :math:`g_2` atoms as a function of lateral
| distance :math:`r` from a centered :math:`g_1` atom.
| PDFPlanar can be used in systems that are inhomogeneous along one axis,
| and homogeneous in a plane.
| In fully homogeneous systems and in the limit of small 'dzheight'
| :math:`\Delta z`, it is the same as the well known three dimensional PDF.
|
| The planar PDF is defined by
|
| .. math::
|
| g_\mathrm{2D}(r) = \left \langle
| \frac{1}{N_{g1}} \cdot \sum_{i}^{N_{g1}} \sum_{j}^{N_{g2}}
| \frac{1}{2 \pi r} \delta(r - r_{ij}) \delta(z_{ij})
| \right \rangle .
|
| where the brackets :math:`\langle \cdot \rangle` denote the ensemble
| average. :math:`\delta(r- r_{ij})` counts the :math:`g_2` atoms at distance
| :math:`r` from atom :math:`i`.
| :math:`\delta(z_{ij})` ensures that only atoms, which lie
| in the same plane :math:`z_i = z_j`, are considered for the PDF.
|
| Discretized for computational purposes the equation reads as
|
| .. math::
|
| g_\mathrm{2D}(r) =
| \frac{1}{N_{g1}} \cdot \sum_{i}^{N_{g1}} \frac{\mathrm{count}\; g_2 \;
| \mathrm{in}\; \Delta V_i(r) }{\Delta V_i(r)} .
|
| where :math:`\Delta V_i(r)` is a ring around atom i, with inner
| radius :math:`r - \frac{\Delta r}{2}`, outer radius
| :math:`r + \frac{\Delta r}{2}` and height :math:`2 \Delta z`.
|
| As the density to normalise the PDF with is unknown, the output is in
| the dimension of number/volume in 1/Å^3.
|
| Functionally, PDFPlanar bins all pairwise :math:`g_1`-:math:`g_2` distances,
| where the z distance is smaller than 'dzheight' in a histogram.
|
| For a more detailed explanation refer to
| :ref:`Explanation: PDF<pdfs-explanation>` and
| :ref:`PDFPlanar Derivation<pdfplanar-derivation>`
|
| Parameters
| ----------
| g1 : MDAnalysis.core.groups.AtomGroup
| First AtomGroup.
| g2 : MDAnalysis.core.groups.AtomGroup
| Second AtomGroup.
| pdf_bin_width : float
| Binwidth of bins in the histogram of the PDF (Å).
| dzheight : float
| dz height of a PDF slab :math:`\Delta z` (Å). :math:`\Delta z` is
| introduced to discretize the delta function :math:`\delta(z_{ij})`.
| It is the maximum :math:`z` distance between atoms which are
| considered to lie in the same plane.
| In the limit of :math:`\Delta z \to 0`, PDFPlanar reaches the
| continous limit. However, if :math:`\Delta z` is too small, there
| are no atoms in ``g2`` to sample.
| We recommend a choice of :math:`\Delta z` that is 1/10th of
| a bond length.
| dmin : float
| Minimum pairwise distance between ``g1`` and ``g2`` (Å).
| dmax : float
| Maximum pairwise distance between ``g1`` and ``g2`` (Å).
| dim : {0, 1, 2}
| Dimension for binning (``x=0``, ``y=1``, ``z=1``).
| zmin : float
| Minimal coordinate for evaluation (in Å) with respect to the center of mass of
| the refgroup.
| If ``zmin=None``, all coordinates down to the lower cell boundary are taken into
| account.
| zmax : float
| Maximal coordinate for evaluation (in Å) with respect to the center of mass of the
| refgroup.
| If ``zmax = None``, all coordinates up to the upper cell boundary are taken into
| account.
| bin_width : float
| Width of the bins (in Å).
| bin_method : {``"com"``, ``"cog"``, ``"coc"``}
| Method for the position binning.
|
| The possible options are center of mass (``"com"``), center of geometry (``"cog"``),
| and center of charge (``"coc"``).
| refgroup : MDAnalysis.core.groups.AtomGroup
| Reference :class:`~MDAnalysis.core.groups.AtomGroup` used for the calculation. If
| ``refgroup`` is provided, the calculation is performed relative to the center of
| mass of the AtomGroup. If ``refgroup`` is :obj:`None` the calculations are performed
| with respect to the center of the (changing) box.
| unwrap : bool
| When :obj:`True`, molecules that are broken due to the periodic boundary conditions
| are made whole.
|
| If the input contains molecules that are already whole, speed up the calculation by
| disabling unwrap. To do so, use the flag ``-no-unwrap`` when using MAICoS from the
| command line, or use ``unwrap=False`` when using MAICoS from the Python interpreter.
|
| Note: Molecules containing virtual sites (e.g. TIP4P water models) are not currently
| supported in MDAnalysis. In this case, you need to provide unwrapped trajectory
| files directly, and disable unwrap. Trajectories can be unwrapped, for example,
| using the ``trjconv`` command of GROMACS.
| pack : bool
| When :obj:`True`, molecules are put back into the unit cell. This is required
| because MAICoS only takes into account molecules that are inside the unit cell.
|
| If the input contains molecules that are already packed, speed up the calculation by
| disabling packing with ``pack=False``.
| jitter : float
| Magnitude of the random noise to add to the atomic positions.
|
| A jitter can be used to stabilize the aliasing effects sometimes appearing when
| histogramming data. The jitter value should be about the precision of the
| trajectory. In that case, using jitter will not alter the results of the histogram.
| If ``jitter = 0.0`` (default), the original atomic positions are kept unchanged.
|
| You can estimate the precision of the positions in your trajectory with
| :func:`maicos.lib.util.trajectory_precision`. Note that if the precision is not the
| same for all frames, the smallest precision should be used.
| concfreq : int
| When concfreq (for conclude frequency) is larger than ``0``, the conclude function
| is called and the output files are written every ``concfreq`` frames.
| output : str
| Output filename.
|
| Attributes
| ----------
| results.bin_pos : numpy.ndarray
| Bin positions (in Å) ranging from ``zmin`` to ``zmax``.
| results.bins: numpy.ndarray
| distances to which the PDF is calculated with shape (pdf_nbins) (Å)
| results.pdf: np.ndrray
| PDF with shape (pdf_nbins, n_bins) (1/Å^3)
|
| Method resolution order:
| PDFPlanar
| maicos.core.planar.PlanarBase
| maicos.core.base.AnalysisBase
| maicos.core.base._Runner
| MDAnalysis.analysis.base.AnalysisBase
| builtins.object
|
| Methods defined here:
|
| __init__(
| self,
| g1: mda.AtomGroup,
| g2: mda.AtomGroup | None = None,
| pdf_bin_width: float = 0.3,
| dzheight: float = 0.1,
| dmin: float = 0.0,
| dmax: float | None = None,
| dim: int = 2,
| zmin: float | None = None,
| zmax: float | None = None,
| bin_width: float = 1,
| bin_method: str = 'com',
| refgroup: mda.AtomGroup | None = None,
| unwrap: bool = False,
| pack: bool = True,
| jitter: float = 0.0,
| concfreq: int = 0,
| output: str = 'pdf.dat'
| ) -> None
| Initialize self. See help(type(self)) for accurate signature.
|
| save(self) -> None
| Save results of analysis to file specified by ``output``.
|
| ----------------------------------------------------------------------
| Readonly properties inherited from maicos.core.planar.PlanarBase:
|
| odims
| Other dimensions perpendicular to dim i.e. (0,2) if dim = 1.
|
| ----------------------------------------------------------------------
| Methods inherited from maicos.core.base.AnalysisBase:
|
| dump(self, filename: str) -> None
| Save analysis state to an ``.npz`` file.
|
| .. warning::
|
| :meth:`dump` is **not** an archival format. The on-disk layout
| tracks the installed MAICoS and MDAnalysis versions and may stop
| loading after an upgrade. Use it to checkpoint or hand off an
| in-progress analysis, not to preserve results long-term. For
| archival output write summary tables with :meth:`save` or export
| :attr:`results` to a stable format.
|
| Persists all statistical accumulators (``means``, ``sems``, ``sums``,
| ``pop``, ``M2``), the ``results`` and ``_obs`` containers, per-frame
| arrays (``timeseries``, ``frames``, ``times``), metadata, the associated
| Universe and the analysed atomgroup. Restore the analysis with :meth:`load`.
|
| Parameters
| ----------
| filename : str
| Path to the output ``.npz`` file.
|
| run(
| self,
| start: int | None = None,
| stop: int | None = None,
| step: int | None = None,
| frames: int | None = None,
| verbose: bool | None = None,
| progressbar_kwargs: dict | None = None
| ) -> Self
| Iterate over the trajectory.
|
| Parameters
| ----------
| start : int
| start frame of analysis
| stop : int
| stop frame of analysis
| step : int
| number of frames to skip between each analysed frame
| frames : array_like
| array of integers or booleans to slice trajectory; ``frames`` can only be
| used *instead* of ``start``, ``stop``, and ``step``. Setting *both*
| ``frames`` and at least one of ``start``, ``stop``, ``step`` to a
| non-default value will raise a :exc:`ValueError`.
| verbose : bool
| Turn on verbosity
| progressbar_kwargs : dict
| ProgressBar keywords with custom parameters regarding progress bar position,
| etc; see :class:`MDAnalysis.lib.log.ProgressBar` for full list.
|
| Returns
| -------
| self : object
| analysis object
|
| savetxt(self, fname: str, X: np.ndarray, columns: list[str] | None = None) -> None
| Save to text.
|
| An extension of the numpy savetxt function. Adds the command line input to the
| header and checks for a doubled defined filesuffix.
|
| Return a header for the text file to save the data to. This method builds a
| generic header that can be used by any MAICoS module. It is called by the save
| method of each module.
|
| The information it collects is:
| - timestamp of the analysis
| - name of the module
| - version of MAICoS that was used
| - command line arguments that were used to run the module
| - module call including the default arguments
| - number of frames that were analyzed
| - atomgroup that was analyzed
| - output messages from modules and base classes (if they exist)
|
| ----------------------------------------------------------------------
| Class methods inherited from maicos.core.base.AnalysisBase:
|
| load(filename: str) -> Self
| Restore an analysis instance from a file created by :meth:`dump`.
|
| Returns a new instance of ``cls`` with all statistical accumulators,
| per-frame arrays, metadata, and the analysed atomgroup populated from
| the file. The rebuilt :class:`MDAnalysis.Universe` carries only the
| topology — no trajectory — so calling :meth:`run` on the returned
| instance raises :class:`RuntimeError`. Use the loaded instance to
| inspect :attr:`results` or to call :meth:`save`.
|
| Parameters
| ----------
| filename : str
| Path to the ``.npz`` file written by :meth:`dump`.
|
| Returns
| -------
| Self
| A new instance of ``cls`` with state restored from ``filename``.
|
| ----------------------------------------------------------------------
| Readonly properties inherited from maicos.core.base.AnalysisBase:
|
| box_center
| Center of the simulation cell.
|
| box_lengths
| Lengths of the simulation cell vectors.
|
| ----------------------------------------------------------------------
| Data descriptors inherited from maicos.core.base._Runner:
|
| __dict__
| dictionary for instance variables
|
| __weakref__
| list of weak references to the object
|
| ----------------------------------------------------------------------
| Class methods inherited from MDAnalysis.analysis.base.AnalysisBase:
|
| get_supported_backends()
| Tuple with backends supported by the core library for a given class.
| User can pass either one of these values as ``backend=...`` to
| :meth:`run()` method, or a custom object that has ``apply`` method
| (see documentation for :meth:`run()`):
|
| - 'serial': no parallelization
| - 'multiprocessing': parallelization using `multiprocessing.Pool`
| - 'dask': parallelization using `dask.delayed.compute()`. Requires
| installation of `mdanalysis[dask]`
|
| If you want to add your own backend to an existing class, pass a
| :class:`backends.BackendBase` subclass (see its documentation to learn
| how to implement it properly), and specify ``unsupported_backend=True``.
|
| Returns
| -------
| tuple
| names of built-in backends that can be used in :meth:`run(backend=...)`
|
|
| .. versionadded:: 2.8.0
|
| ----------------------------------------------------------------------
| Readonly properties inherited from MDAnalysis.analysis.base.AnalysisBase:
|
| parallelizable
| Boolean mark showing that a given class can be parallelizable with
| split-apply-combine procedure. Namely, if we can safely distribute
| :meth:`_single_frame` to multiple workers and then combine them with a
| proper :meth:`_conclude` call. If set to ``False``, no backends except
| for ``serial`` are supported.
|
| .. note:: If you want to check parallelizability of the whole class, without
| explicitly creating an instance of the class, see
| :attr:`_analysis_algorithm_is_parallelizable`. Note that you
| setting it to other value will break things if the algorithm
| behind the analysis is not trivially parallelizable.
|
|
| Returns
| -------
| bool
| if a given ``AnalysisBase`` subclass instance
| is parallelizable with split-apply-combine, or not
|
|
| .. versionadded:: 2.8.0
class RDFDiporder(builtins.object)
| RDFDiporder(
| g1: mda.AtomGroup,
| g2: mda.AtomGroup | None = None,
| norm: str = 'rdf',
| rmin: float = 0.0,
| rmax: float = 15.0,
| bin_width: float = 0.1,
| bin_method: str = 'com',
| grouping: str = 'residues',
| refgroup: mda.AtomGroup | None = None,
| unwrap: bool = True,
| pack: bool = True,
| jitter: float = 0.0,
| concfreq: int = 0,
| output: str = 'diporderrdf.dat'
| ) -> None
|
| Class has been moved to the ``scatterkit`` package.
|
| Methods defined here:
|
| __init__(
| self,
| g1: mda.AtomGroup,
| g2: mda.AtomGroup | None = None,
| norm: str = 'rdf',
| rmin: float = 0.0,
| rmax: float = 15.0,
| bin_width: float = 0.1,
| bin_method: str = 'com',
| grouping: str = 'residues',
| refgroup: mda.AtomGroup | None = None,
| unwrap: bool = True,
| pack: bool = True,
| jitter: float = 0.0,
| concfreq: int = 0,
| output: str = 'diporderrdf.dat'
| ) -> None
| Initialize self. See help(type(self)) for accurate signature.
|
| ----------------------------------------------------------------------
| Data descriptors defined here:
|
| __dict__
| dictionary for instance variables
|
| __weakref__
| list of weak references to the object
class Saxs(builtins.object)
| Saxs(
| atomgroup: mda.AtomGroup,
| bin_spectrum: bool = True,
| qmin: float = 0,
| qmax: float = 6,
| dq: float = 0.1,
| thetamin: float = 0,
| thetamax: float = 180,
| refgroup: mda.AtomGroup | None = None,
| unwrap: bool = False,
| pack: bool = True,
| jitter: float = 0.0,
| concfreq: int = 0,
| output: str = 'sq.dat'
| ) -> None
|
| Class has been moved to the ``scatterkit`` package.
|
| Methods defined here:
|
| __init__(
| self,
| atomgroup: mda.AtomGroup,
| bin_spectrum: bool = True,
| qmin: float = 0,
| qmax: float = 6,
| dq: float = 0.1,
| thetamin: float = 0,
| thetamax: float = 180,
| refgroup: mda.AtomGroup | None = None,
| unwrap: bool = False,
| pack: bool = True,
| jitter: float = 0.0,
| concfreq: int = 0,
| output: str = 'sq.dat'
| ) -> None
| Initialize self. See help(type(self)) for accurate signature.
|
| ----------------------------------------------------------------------
| Data descriptors defined here:
|
| __dict__
| dictionary for instance variables
|
| __weakref__
| list of weak references to the object
class TemperaturePlanar(maicos.core.planar.ProfilePlanarBase)
| TemperaturePlanar(
| atomgroup: mda.AtomGroup,
| dim: int = 2,
| zmin: float | None = None,
| zmax: float | None = None,
| bin_width: float = 1,
| bin_method: str = 'com',
| grouping: str = 'atoms',
| sym: bool = False,
| refgroup: mda.AtomGroup | None = None,
| unwrap: bool = True,
| pack: bool = True,
| jitter: float = 0.0,
| concfreq: int = 0,
| output: str = 'temperature.dat'
| ) -> None
|
| Temperature profiles in a cartesian geometry.
|
| Currently only atomistic temperature profiles are supported. Therefore grouping per
| molecule, segment, residue, or fragment is not possible.
|
| For the correlation analysis the central bin
| (:math:`N / 2`) of the 0th's group profile is used. For further information on the correlation analysis please
| refer to :class:`AnalysisBase <maicos.core.base.AnalysisBase>` or the
| :ref:`general-design` section.
|
| Parameters
| ----------
| atomgroup : MDAnalysis.core.groups.AtomGroup
| A :class:`~MDAnalysis.core.groups.AtomGroup` for which the calculations are
| performed.
|
| dim : {0, 1, 2}
| Dimension for binning (``x=0``, ``y=1``, ``z=1``).
| zmin : float
| Minimal coordinate for evaluation (in Å) with respect to the center of mass of
| the refgroup.
| If ``zmin=None``, all coordinates down to the lower cell boundary are taken into
| account.
| zmax : float
| Maximal coordinate for evaluation (in Å) with respect to the center of mass of the
| refgroup.
| If ``zmax = None``, all coordinates up to the upper cell boundary are taken into
| account.
| bin_width : float
| Width of the bins (in Å).
| bin_method : {``"com"``, ``"cog"``, ``"coc"``}
| Method for the position binning.
|
| The possible options are center of mass (``"com"``), center of geometry (``"cog"``),
| and center of charge (``"coc"``).
| grouping : {``"atoms"``, ``"residues"``, ``"segments"``, ``"molecules"``, ``"fragments"``}
| Atom grouping for the calculations.
|
| The possible grouping options are the atom positions (in the case where
| ``grouping="atoms"``) or the center of mass of the specified grouping unit (in the
| case where ``grouping="residues"``, ``"segments"``, ``"molecules"`` or
| ``"fragments"``).
| sym : bool
| Symmetrize the profile. Only works in combination with ``refgroup``.
| refgroup : MDAnalysis.core.groups.AtomGroup
| Reference :class:`~MDAnalysis.core.groups.AtomGroup` used for the calculation. If
| ``refgroup`` is provided, the calculation is performed relative to the center of
| mass of the AtomGroup. If ``refgroup`` is :obj:`None` the calculations are performed
| with respect to the center of the (changing) box.
| unwrap : bool
| When :obj:`True`, molecules that are broken due to the periodic boundary conditions
| are made whole.
|
| If the input contains molecules that are already whole, speed up the calculation by
| disabling unwrap. To do so, use the flag ``-no-unwrap`` when using MAICoS from the
| command line, or use ``unwrap=False`` when using MAICoS from the Python interpreter.
|
| Note: Molecules containing virtual sites (e.g. TIP4P water models) are not currently
| supported in MDAnalysis. In this case, you need to provide unwrapped trajectory
| files directly, and disable unwrap. Trajectories can be unwrapped, for example,
| using the ``trjconv`` command of GROMACS.
| pack : bool
| When :obj:`True`, molecules are put back into the unit cell. This is required
| because MAICoS only takes into account molecules that are inside the unit cell.
|
| If the input contains molecules that are already packed, speed up the calculation by
| disabling packing with ``pack=False``.
| jitter : float
| Magnitude of the random noise to add to the atomic positions.
|
| A jitter can be used to stabilize the aliasing effects sometimes appearing when
| histogramming data. The jitter value should be about the precision of the
| trajectory. In that case, using jitter will not alter the results of the histogram.
| If ``jitter = 0.0`` (default), the original atomic positions are kept unchanged.
|
| You can estimate the precision of the positions in your trajectory with
| :func:`maicos.lib.util.trajectory_precision`. Note that if the precision is not the
| same for all frames, the smallest precision should be used.
| concfreq : int
| When concfreq (for conclude frequency) is larger than ``0``, the conclude function
| is called and the output files are written every ``concfreq`` frames.
| output : str
| Output filename.
|
| Attributes
| ----------
| results.bin_pos : numpy.ndarray
| Bin positions (in Å) ranging from ``zmin`` to ``zmax``.
| results.profile : numpy.ndarray
| Calculated profile.
| results.dprofile : numpy.ndarray
| Estimated profile's uncertainity.
|
| Method resolution order:
| TemperaturePlanar
| maicos.core.planar.ProfilePlanarBase
| maicos.core.planar.PlanarBase
| maicos.core.base.AnalysisBase
| maicos.core.base._Runner
| MDAnalysis.analysis.base.AnalysisBase
| maicos.core.base.ProfileBase
| builtins.object
|
| Methods defined here:
|
| __init__(
| self,
| atomgroup: mda.AtomGroup,
| dim: int = 2,
| zmin: float | None = None,
| zmax: float | None = None,
| bin_width: float = 1,
| bin_method: str = 'com',
| grouping: str = 'atoms',
| sym: bool = False,
| refgroup: mda.AtomGroup | None = None,
| unwrap: bool = True,
| pack: bool = True,
| jitter: float = 0.0,
| concfreq: int = 0,
| output: str = 'temperature.dat'
| ) -> None
| Initialize self. See help(type(self)) for accurate signature.
|
| ----------------------------------------------------------------------
| Readonly properties inherited from maicos.core.planar.PlanarBase:
|
| odims
| Other dimensions perpendicular to dim i.e. (0,2) if dim = 1.
|
| ----------------------------------------------------------------------
| Methods inherited from maicos.core.base.AnalysisBase:
|
| dump(self, filename: str) -> None
| Save analysis state to an ``.npz`` file.
|
| .. warning::
|
| :meth:`dump` is **not** an archival format. The on-disk layout
| tracks the installed MAICoS and MDAnalysis versions and may stop
| loading after an upgrade. Use it to checkpoint or hand off an
| in-progress analysis, not to preserve results long-term. For
| archival output write summary tables with :meth:`save` or export
| :attr:`results` to a stable format.
|
| Persists all statistical accumulators (``means``, ``sems``, ``sums``,
| ``pop``, ``M2``), the ``results`` and ``_obs`` containers, per-frame
| arrays (``timeseries``, ``frames``, ``times``), metadata, the associated
| Universe and the analysed atomgroup. Restore the analysis with :meth:`load`.
|
| Parameters
| ----------
| filename : str
| Path to the output ``.npz`` file.
|
| run(
| self,
| start: int | None = None,
| stop: int | None = None,
| step: int | None = None,
| frames: int | None = None,
| verbose: bool | None = None,
| progressbar_kwargs: dict | None = None
| ) -> Self
| Iterate over the trajectory.
|
| Parameters
| ----------
| start : int
| start frame of analysis
| stop : int
| stop frame of analysis
| step : int
| number of frames to skip between each analysed frame
| frames : array_like
| array of integers or booleans to slice trajectory; ``frames`` can only be
| used *instead* of ``start``, ``stop``, and ``step``. Setting *both*
| ``frames`` and at least one of ``start``, ``stop``, ``step`` to a
| non-default value will raise a :exc:`ValueError`.
| verbose : bool
| Turn on verbosity
| progressbar_kwargs : dict
| ProgressBar keywords with custom parameters regarding progress bar position,
| etc; see :class:`MDAnalysis.lib.log.ProgressBar` for full list.
|
| Returns
| -------
| self : object
| analysis object
|
| savetxt(self, fname: str, X: np.ndarray, columns: list[str] | None = None) -> None
| Save to text.
|
| An extension of the numpy savetxt function. Adds the command line input to the
| header and checks for a doubled defined filesuffix.
|
| Return a header for the text file to save the data to. This method builds a
| generic header that can be used by any MAICoS module. It is called by the save
| method of each module.
|
| The information it collects is:
| - timestamp of the analysis
| - name of the module
| - version of MAICoS that was used
| - command line arguments that were used to run the module
| - module call including the default arguments
| - number of frames that were analyzed
| - atomgroup that was analyzed
| - output messages from modules and base classes (if they exist)
|
| ----------------------------------------------------------------------
| Class methods inherited from maicos.core.base.AnalysisBase:
|
| load(filename: str) -> Self
| Restore an analysis instance from a file created by :meth:`dump`.
|
| Returns a new instance of ``cls`` with all statistical accumulators,
| per-frame arrays, metadata, and the analysed atomgroup populated from
| the file. The rebuilt :class:`MDAnalysis.Universe` carries only the
| topology — no trajectory — so calling :meth:`run` on the returned
| instance raises :class:`RuntimeError`. Use the loaded instance to
| inspect :attr:`results` or to call :meth:`save`.
|
| Parameters
| ----------
| filename : str
| Path to the ``.npz`` file written by :meth:`dump`.
|
| Returns
| -------
| Self
| A new instance of ``cls`` with state restored from ``filename``.
|
| ----------------------------------------------------------------------
| Readonly properties inherited from maicos.core.base.AnalysisBase:
|
| box_center
| Center of the simulation cell.
|
| box_lengths
| Lengths of the simulation cell vectors.
|
| ----------------------------------------------------------------------
| Data descriptors inherited from maicos.core.base._Runner:
|
| __dict__
| dictionary for instance variables
|
| __weakref__
| list of weak references to the object
|
| ----------------------------------------------------------------------
| Class methods inherited from MDAnalysis.analysis.base.AnalysisBase:
|
| get_supported_backends()
| Tuple with backends supported by the core library for a given class.
| User can pass either one of these values as ``backend=...`` to
| :meth:`run()` method, or a custom object that has ``apply`` method
| (see documentation for :meth:`run()`):
|
| - 'serial': no parallelization
| - 'multiprocessing': parallelization using `multiprocessing.Pool`
| - 'dask': parallelization using `dask.delayed.compute()`. Requires
| installation of `mdanalysis[dask]`
|
| If you want to add your own backend to an existing class, pass a
| :class:`backends.BackendBase` subclass (see its documentation to learn
| how to implement it properly), and specify ``unsupported_backend=True``.
|
| Returns
| -------
| tuple
| names of built-in backends that can be used in :meth:`run(backend=...)`
|
|
| .. versionadded:: 2.8.0
|
| ----------------------------------------------------------------------
| Readonly properties inherited from MDAnalysis.analysis.base.AnalysisBase:
|
| parallelizable
| Boolean mark showing that a given class can be parallelizable with
| split-apply-combine procedure. Namely, if we can safely distribute
| :meth:`_single_frame` to multiple workers and then combine them with a
| proper :meth:`_conclude` call. If set to ``False``, no backends except
| for ``serial`` are supported.
|
| .. note:: If you want to check parallelizability of the whole class, without
| explicitly creating an instance of the class, see
| :attr:`_analysis_algorithm_is_parallelizable`. Note that you
| setting it to other value will break things if the algorithm
| behind the analysis is not trivially parallelizable.
|
|
| Returns
| -------
| bool
| if a given ``AnalysisBase`` subclass instance
| is parallelizable with split-apply-combine, or not
|
|
| .. versionadded:: 2.8.0
|
| ----------------------------------------------------------------------
| Methods inherited from maicos.core.base.ProfileBase:
|
| save(self) -> None
| Save results of analysis to file specified by ``output``.
class VelocityCylinder(maicos.core.cylinder.ProfileCylinderBase)
| VelocityCylinder(
| atomgroup: mda.AtomGroup,
| vdim: int = 0,
| flux: bool = False,
| dim: int = 2,
| zmin: float | None = None,
| zmax: float | None = None,
| rmin: float = 0,
| rmax: float | None = None,
| bin_width: int = 1,
| bin_method: str = 'com',
| grouping: str = 'atoms',
| refgroup: mda.AtomGroup | None = None,
| unwrap: bool = True,
| pack: bool = True,
| jitter: float = 0.0,
| concfreq: int = 0,
| output: str = 'velocity.dat'
| ) -> None
|
| Cartesian velocity profile across a cylinder.
|
| Reads in coordinates and velocities from a trajectory and calculates a velocity
| :math:`[\mathrm{Å/ps}]` or a flux per unit area :math:`[\mathrm{Å^{-2}\,ps^{-1}}]`
| profile along a given axis.
|
| The ``grouping`` keyword gives you fine control over the velocity profile, e.g. you
| can choose atomar or molecular velocities. Note that if the first one is employed
| for complex compounds, usually a contribution corresponding to the vorticity appears
| in the profile.
|
| For the correlation analysis the 0th bin of the 0th's
| group profile is used. For further information on the correlation analysis please
| refer to :class:`AnalysisBase <maicos.core.base.AnalysisBase>` or the
| :ref:`general-design` section.
|
| Parameters
| ----------
| atomgroup : MDAnalysis.core.groups.AtomGroup
| A :class:`~MDAnalysis.core.groups.AtomGroup` for which the calculations are
| performed.
| vdim : {0, 1, 2}
| Dimension for velocity binning (``x=0``, ``y=1``, ``z=1``).
| flux : bool
| Calculate the flux (:math:`[Å^2/\mathrm{ps}]`) instead of the velocity.
| dim : {0, 1, 2}
| Dimension for binning (``x=0``, ``y=1``, ``z=1``).
| zmin : float
| Minimal coordinate for evaluation (in Å) with respect to the center of mass of
| the refgroup.
| If ``zmin=None``, all coordinates down to the lower cell boundary are taken into
| account.
| zmax : float
| Maximal coordinate for evaluation (in Å) with respect to the center of mass of the
| refgroup.
| If ``zmax = None``, all coordinates up to the upper cell boundary are taken into
| account.
| rmin : float
| Minimal radial coordinate relative to the center of mass of the refgroup for
| evaluation (in Å).
| rmax : float
| Maximal radial coordinate relative to the center of mass of the refgroup for
| evaluation (in Å).
|
| If ``rmax=None``, the box extension is taken.
| bin_width : float
| Width of the bins (in Å).
| bin_method : {``"com"``, ``"cog"``, ``"coc"``}
| Method for the position binning.
|
| The possible options are center of mass (``"com"``), center of geometry (``"cog"``),
| and center of charge (``"coc"``).
| grouping : {``"atoms"``, ``"residues"``, ``"segments"``, ``"molecules"``, ``"fragments"``}
| Atom grouping for the calculations.
|
| The possible grouping options are the atom positions (in the case where
| ``grouping="atoms"``) or the center of mass of the specified grouping unit (in the
| case where ``grouping="residues"``, ``"segments"``, ``"molecules"`` or
| ``"fragments"``).
| refgroup : MDAnalysis.core.groups.AtomGroup
| Reference :class:`~MDAnalysis.core.groups.AtomGroup` used for the calculation. If
| ``refgroup`` is provided, the calculation is performed relative to the center of
| mass of the AtomGroup. If ``refgroup`` is :obj:`None` the calculations are performed
| with respect to the center of the (changing) box.
| unwrap : bool
| When :obj:`True`, molecules that are broken due to the periodic boundary conditions
| are made whole.
|
| If the input contains molecules that are already whole, speed up the calculation by
| disabling unwrap. To do so, use the flag ``-no-unwrap`` when using MAICoS from the
| command line, or use ``unwrap=False`` when using MAICoS from the Python interpreter.
|
| Note: Molecules containing virtual sites (e.g. TIP4P water models) are not currently
| supported in MDAnalysis. In this case, you need to provide unwrapped trajectory
| files directly, and disable unwrap. Trajectories can be unwrapped, for example,
| using the ``trjconv`` command of GROMACS.
| pack : bool
| When :obj:`True`, molecules are put back into the unit cell. This is required
| because MAICoS only takes into account molecules that are inside the unit cell.
|
| If the input contains molecules that are already packed, speed up the calculation by
| disabling packing with ``pack=False``.
| jitter : float
| Magnitude of the random noise to add to the atomic positions.
|
| A jitter can be used to stabilize the aliasing effects sometimes appearing when
| histogramming data. The jitter value should be about the precision of the
| trajectory. In that case, using jitter will not alter the results of the histogram.
| If ``jitter = 0.0`` (default), the original atomic positions are kept unchanged.
|
| You can estimate the precision of the positions in your trajectory with
| :func:`maicos.lib.util.trajectory_precision`. Note that if the precision is not the
| same for all frames, the smallest precision should be used.
| concfreq : int
| When concfreq (for conclude frequency) is larger than ``0``, the conclude function
| is called and the output files are written every ``concfreq`` frames.
| output : str
| Output filename.
|
| Attributes
| ----------
| results.bin_pos : numpy.ndarray
| Bin positions (in Å) ranging from ``rmin`` to ``rmax``.
| results.profile : numpy.ndarray
| Calculated profile.
| results.dprofile : numpy.ndarray
| Estimated profile's uncertainity.
|
| Method resolution order:
| VelocityCylinder
| maicos.core.cylinder.ProfileCylinderBase
| maicos.core.cylinder.CylinderBase
| maicos.core.planar.PlanarBase
| maicos.core.base.AnalysisBase
| maicos.core.base._Runner
| MDAnalysis.analysis.base.AnalysisBase
| maicos.core.base.ProfileBase
| builtins.object
|
| Methods defined here:
|
| __init__(
| self,
| atomgroup: mda.AtomGroup,
| vdim: int = 0,
| flux: bool = False,
| dim: int = 2,
| zmin: float | None = None,
| zmax: float | None = None,
| rmin: float = 0,
| rmax: float | None = None,
| bin_width: int = 1,
| bin_method: str = 'com',
| grouping: str = 'atoms',
| refgroup: mda.AtomGroup | None = None,
| unwrap: bool = True,
| pack: bool = True,
| jitter: float = 0.0,
| concfreq: int = 0,
| output: str = 'velocity.dat'
| ) -> None
| Initialize self. See help(type(self)) for accurate signature.
|
| ----------------------------------------------------------------------
| Readonly properties inherited from maicos.core.planar.PlanarBase:
|
| odims
| Other dimensions perpendicular to dim i.e. (0,2) if dim = 1.
|
| ----------------------------------------------------------------------
| Methods inherited from maicos.core.base.AnalysisBase:
|
| dump(self, filename: str) -> None
| Save analysis state to an ``.npz`` file.
|
| .. warning::
|
| :meth:`dump` is **not** an archival format. The on-disk layout
| tracks the installed MAICoS and MDAnalysis versions and may stop
| loading after an upgrade. Use it to checkpoint or hand off an
| in-progress analysis, not to preserve results long-term. For
| archival output write summary tables with :meth:`save` or export
| :attr:`results` to a stable format.
|
| Persists all statistical accumulators (``means``, ``sems``, ``sums``,
| ``pop``, ``M2``), the ``results`` and ``_obs`` containers, per-frame
| arrays (``timeseries``, ``frames``, ``times``), metadata, the associated
| Universe and the analysed atomgroup. Restore the analysis with :meth:`load`.
|
| Parameters
| ----------
| filename : str
| Path to the output ``.npz`` file.
|
| run(
| self,
| start: int | None = None,
| stop: int | None = None,
| step: int | None = None,
| frames: int | None = None,
| verbose: bool | None = None,
| progressbar_kwargs: dict | None = None
| ) -> Self
| Iterate over the trajectory.
|
| Parameters
| ----------
| start : int
| start frame of analysis
| stop : int
| stop frame of analysis
| step : int
| number of frames to skip between each analysed frame
| frames : array_like
| array of integers or booleans to slice trajectory; ``frames`` can only be
| used *instead* of ``start``, ``stop``, and ``step``. Setting *both*
| ``frames`` and at least one of ``start``, ``stop``, ``step`` to a
| non-default value will raise a :exc:`ValueError`.
| verbose : bool
| Turn on verbosity
| progressbar_kwargs : dict
| ProgressBar keywords with custom parameters regarding progress bar position,
| etc; see :class:`MDAnalysis.lib.log.ProgressBar` for full list.
|
| Returns
| -------
| self : object
| analysis object
|
| savetxt(self, fname: str, X: np.ndarray, columns: list[str] | None = None) -> None
| Save to text.
|
| An extension of the numpy savetxt function. Adds the command line input to the
| header and checks for a doubled defined filesuffix.
|
| Return a header for the text file to save the data to. This method builds a
| generic header that can be used by any MAICoS module. It is called by the save
| method of each module.
|
| The information it collects is:
| - timestamp of the analysis
| - name of the module
| - version of MAICoS that was used
| - command line arguments that were used to run the module
| - module call including the default arguments
| - number of frames that were analyzed
| - atomgroup that was analyzed
| - output messages from modules and base classes (if they exist)
|
| ----------------------------------------------------------------------
| Class methods inherited from maicos.core.base.AnalysisBase:
|
| load(filename: str) -> Self
| Restore an analysis instance from a file created by :meth:`dump`.
|
| Returns a new instance of ``cls`` with all statistical accumulators,
| per-frame arrays, metadata, and the analysed atomgroup populated from
| the file. The rebuilt :class:`MDAnalysis.Universe` carries only the
| topology — no trajectory — so calling :meth:`run` on the returned
| instance raises :class:`RuntimeError`. Use the loaded instance to
| inspect :attr:`results` or to call :meth:`save`.
|
| Parameters
| ----------
| filename : str
| Path to the ``.npz`` file written by :meth:`dump`.
|
| Returns
| -------
| Self
| A new instance of ``cls`` with state restored from ``filename``.
|
| ----------------------------------------------------------------------
| Readonly properties inherited from maicos.core.base.AnalysisBase:
|
| box_center
| Center of the simulation cell.
|
| box_lengths
| Lengths of the simulation cell vectors.
|
| ----------------------------------------------------------------------
| Data descriptors inherited from maicos.core.base._Runner:
|
| __dict__
| dictionary for instance variables
|
| __weakref__
| list of weak references to the object
|
| ----------------------------------------------------------------------
| Class methods inherited from MDAnalysis.analysis.base.AnalysisBase:
|
| get_supported_backends()
| Tuple with backends supported by the core library for a given class.
| User can pass either one of these values as ``backend=...`` to
| :meth:`run()` method, or a custom object that has ``apply`` method
| (see documentation for :meth:`run()`):
|
| - 'serial': no parallelization
| - 'multiprocessing': parallelization using `multiprocessing.Pool`
| - 'dask': parallelization using `dask.delayed.compute()`. Requires
| installation of `mdanalysis[dask]`
|
| If you want to add your own backend to an existing class, pass a
| :class:`backends.BackendBase` subclass (see its documentation to learn
| how to implement it properly), and specify ``unsupported_backend=True``.
|
| Returns
| -------
| tuple
| names of built-in backends that can be used in :meth:`run(backend=...)`
|
|
| .. versionadded:: 2.8.0
|
| ----------------------------------------------------------------------
| Readonly properties inherited from MDAnalysis.analysis.base.AnalysisBase:
|
| parallelizable
| Boolean mark showing that a given class can be parallelizable with
| split-apply-combine procedure. Namely, if we can safely distribute
| :meth:`_single_frame` to multiple workers and then combine them with a
| proper :meth:`_conclude` call. If set to ``False``, no backends except
| for ``serial`` are supported.
|
| .. note:: If you want to check parallelizability of the whole class, without
| explicitly creating an instance of the class, see
| :attr:`_analysis_algorithm_is_parallelizable`. Note that you
| setting it to other value will break things if the algorithm
| behind the analysis is not trivially parallelizable.
|
|
| Returns
| -------
| bool
| if a given ``AnalysisBase`` subclass instance
| is parallelizable with split-apply-combine, or not
|
|
| .. versionadded:: 2.8.0
|
| ----------------------------------------------------------------------
| Methods inherited from maicos.core.base.ProfileBase:
|
| save(self) -> None
| Save results of analysis to file specified by ``output``.
class VelocityPlanar(maicos.core.planar.ProfilePlanarBase)
| VelocityPlanar(
| atomgroup: mda.AtomGroup,
| sym_odd: bool = False,
| vdim: int = 0,
| flux: bool = False,
| dim: int = 2,
| zmin: float | None = None,
| zmax: float | None = None,
| bin_width: float = 1.0,
| bin_method: str = 'com',
| grouping: str = 'atoms',
| sym: bool = False,
| refgroup: mda.AtomGroup | None = None,
| unwrap: bool = True,
| pack: bool = True,
| jitter: float = 0.0,
| concfreq: int = 0,
| output: str = 'velocity.dat'
| ) -> None
|
| Velocity profiles in a cartesian geometry.
|
| Reads in coordinates and velocities from a trajectory and calculates a velocity
| :math:`[\mathrm{Å/ps}]` or a flux per unit area :math:`[\mathrm{Å^{-2}\,ps^{-1}}]`
| profile along a given axis.
|
| The ``grouping`` keyword gives you fine control over the velocity profile, e.g. you
| can choose atomar or molecular velocities. Note that if the first one is employed
| for complex compounds, usually a contribution corresponding to the vorticity appears
| in the profile.
|
| For the correlation analysis the central bin
| (:math:`N / 2`) of the 0th's group profile is used. For further information on the correlation analysis please
| refer to :class:`AnalysisBase <maicos.core.base.AnalysisBase>` or the
| :ref:`general-design` section.
|
| Parameters
| ----------
| atomgroup : MDAnalysis.core.groups.AtomGroup
| A :class:`~MDAnalysis.core.groups.AtomGroup` for which the calculations are
| performed.
| sym_odd : bool,
| Parity of the profile. If :obj:`False`, the profile will be symmetrized. If
| :obj:`True`, the profile is antisymmetrized. Only relevant in combination with
| ``sym``.
| vdim : {0, 1, 2}
| Dimension for velocity binning (``x=0``, ``y=1``, ``z=1``).
| flux : bool
| Calculate the flux (:math:`[Å^2/\mathrm{ps}]`) instead of the velocity.
|
| dim : {0, 1, 2}
| Dimension for binning (``x=0``, ``y=1``, ``z=1``).
| zmin : float
| Minimal coordinate for evaluation (in Å) with respect to the center of mass of
| the refgroup.
| If ``zmin=None``, all coordinates down to the lower cell boundary are taken into
| account.
| zmax : float
| Maximal coordinate for evaluation (in Å) with respect to the center of mass of the
| refgroup.
| If ``zmax = None``, all coordinates up to the upper cell boundary are taken into
| account.
| bin_width : float
| Width of the bins (in Å).
| bin_method : {``"com"``, ``"cog"``, ``"coc"``}
| Method for the position binning.
|
| The possible options are center of mass (``"com"``), center of geometry (``"cog"``),
| and center of charge (``"coc"``).
| grouping : {``"atoms"``, ``"residues"``, ``"segments"``, ``"molecules"``, ``"fragments"``}
| Atom grouping for the calculations.
|
| The possible grouping options are the atom positions (in the case where
| ``grouping="atoms"``) or the center of mass of the specified grouping unit (in the
| case where ``grouping="residues"``, ``"segments"``, ``"molecules"`` or
| ``"fragments"``).
| sym : bool
| Symmetrize the profile. Only works in combination with ``refgroup``.
| refgroup : MDAnalysis.core.groups.AtomGroup
| Reference :class:`~MDAnalysis.core.groups.AtomGroup` used for the calculation. If
| ``refgroup`` is provided, the calculation is performed relative to the center of
| mass of the AtomGroup. If ``refgroup`` is :obj:`None` the calculations are performed
| with respect to the center of the (changing) box.
| unwrap : bool
| When :obj:`True`, molecules that are broken due to the periodic boundary conditions
| are made whole.
|
| If the input contains molecules that are already whole, speed up the calculation by
| disabling unwrap. To do so, use the flag ``-no-unwrap`` when using MAICoS from the
| command line, or use ``unwrap=False`` when using MAICoS from the Python interpreter.
|
| Note: Molecules containing virtual sites (e.g. TIP4P water models) are not currently
| supported in MDAnalysis. In this case, you need to provide unwrapped trajectory
| files directly, and disable unwrap. Trajectories can be unwrapped, for example,
| using the ``trjconv`` command of GROMACS.
| pack : bool
| When :obj:`True`, molecules are put back into the unit cell. This is required
| because MAICoS only takes into account molecules that are inside the unit cell.
|
| If the input contains molecules that are already packed, speed up the calculation by
| disabling packing with ``pack=False``.
| jitter : float
| Magnitude of the random noise to add to the atomic positions.
|
| A jitter can be used to stabilize the aliasing effects sometimes appearing when
| histogramming data. The jitter value should be about the precision of the
| trajectory. In that case, using jitter will not alter the results of the histogram.
| If ``jitter = 0.0`` (default), the original atomic positions are kept unchanged.
|
| You can estimate the precision of the positions in your trajectory with
| :func:`maicos.lib.util.trajectory_precision`. Note that if the precision is not the
| same for all frames, the smallest precision should be used.
| concfreq : int
| When concfreq (for conclude frequency) is larger than ``0``, the conclude function
| is called and the output files are written every ``concfreq`` frames.
| output : str
| Output filename.
|
| Attributes
| ----------
| results.bin_pos : numpy.ndarray
| Bin positions (in Å) ranging from ``zmin`` to ``zmax``.
| results.profile : numpy.ndarray
| Calculated profile.
| results.dprofile : numpy.ndarray
| Estimated profile's uncertainity.
|
| Method resolution order:
| VelocityPlanar
| maicos.core.planar.ProfilePlanarBase
| maicos.core.planar.PlanarBase
| maicos.core.base.AnalysisBase
| maicos.core.base._Runner
| MDAnalysis.analysis.base.AnalysisBase
| maicos.core.base.ProfileBase
| builtins.object
|
| Methods defined here:
|
| __init__(
| self,
| atomgroup: mda.AtomGroup,
| sym_odd: bool = False,
| vdim: int = 0,
| flux: bool = False,
| dim: int = 2,
| zmin: float | None = None,
| zmax: float | None = None,
| bin_width: float = 1.0,
| bin_method: str = 'com',
| grouping: str = 'atoms',
| sym: bool = False,
| refgroup: mda.AtomGroup | None = None,
| unwrap: bool = True,
| pack: bool = True,
| jitter: float = 0.0,
| concfreq: int = 0,
| output: str = 'velocity.dat'
| ) -> None
| Initialize self. See help(type(self)) for accurate signature.
|
| ----------------------------------------------------------------------
| Readonly properties inherited from maicos.core.planar.PlanarBase:
|
| odims
| Other dimensions perpendicular to dim i.e. (0,2) if dim = 1.
|
| ----------------------------------------------------------------------
| Methods inherited from maicos.core.base.AnalysisBase:
|
| dump(self, filename: str) -> None
| Save analysis state to an ``.npz`` file.
|
| .. warning::
|
| :meth:`dump` is **not** an archival format. The on-disk layout
| tracks the installed MAICoS and MDAnalysis versions and may stop
| loading after an upgrade. Use it to checkpoint or hand off an
| in-progress analysis, not to preserve results long-term. For
| archival output write summary tables with :meth:`save` or export
| :attr:`results` to a stable format.
|
| Persists all statistical accumulators (``means``, ``sems``, ``sums``,
| ``pop``, ``M2``), the ``results`` and ``_obs`` containers, per-frame
| arrays (``timeseries``, ``frames``, ``times``), metadata, the associated
| Universe and the analysed atomgroup. Restore the analysis with :meth:`load`.
|
| Parameters
| ----------
| filename : str
| Path to the output ``.npz`` file.
|
| run(
| self,
| start: int | None = None,
| stop: int | None = None,
| step: int | None = None,
| frames: int | None = None,
| verbose: bool | None = None,
| progressbar_kwargs: dict | None = None
| ) -> Self
| Iterate over the trajectory.
|
| Parameters
| ----------
| start : int
| start frame of analysis
| stop : int
| stop frame of analysis
| step : int
| number of frames to skip between each analysed frame
| frames : array_like
| array of integers or booleans to slice trajectory; ``frames`` can only be
| used *instead* of ``start``, ``stop``, and ``step``. Setting *both*
| ``frames`` and at least one of ``start``, ``stop``, ``step`` to a
| non-default value will raise a :exc:`ValueError`.
| verbose : bool
| Turn on verbosity
| progressbar_kwargs : dict
| ProgressBar keywords with custom parameters regarding progress bar position,
| etc; see :class:`MDAnalysis.lib.log.ProgressBar` for full list.
|
| Returns
| -------
| self : object
| analysis object
|
| savetxt(self, fname: str, X: np.ndarray, columns: list[str] | None = None) -> None
| Save to text.
|
| An extension of the numpy savetxt function. Adds the command line input to the
| header and checks for a doubled defined filesuffix.
|
| Return a header for the text file to save the data to. This method builds a
| generic header that can be used by any MAICoS module. It is called by the save
| method of each module.
|
| The information it collects is:
| - timestamp of the analysis
| - name of the module
| - version of MAICoS that was used
| - command line arguments that were used to run the module
| - module call including the default arguments
| - number of frames that were analyzed
| - atomgroup that was analyzed
| - output messages from modules and base classes (if they exist)
|
| ----------------------------------------------------------------------
| Class methods inherited from maicos.core.base.AnalysisBase:
|
| load(filename: str) -> Self
| Restore an analysis instance from a file created by :meth:`dump`.
|
| Returns a new instance of ``cls`` with all statistical accumulators,
| per-frame arrays, metadata, and the analysed atomgroup populated from
| the file. The rebuilt :class:`MDAnalysis.Universe` carries only the
| topology — no trajectory — so calling :meth:`run` on the returned
| instance raises :class:`RuntimeError`. Use the loaded instance to
| inspect :attr:`results` or to call :meth:`save`.
|
| Parameters
| ----------
| filename : str
| Path to the ``.npz`` file written by :meth:`dump`.
|
| Returns
| -------
| Self
| A new instance of ``cls`` with state restored from ``filename``.
|
| ----------------------------------------------------------------------
| Readonly properties inherited from maicos.core.base.AnalysisBase:
|
| box_center
| Center of the simulation cell.
|
| box_lengths
| Lengths of the simulation cell vectors.
|
| ----------------------------------------------------------------------
| Data descriptors inherited from maicos.core.base._Runner:
|
| __dict__
| dictionary for instance variables
|
| __weakref__
| list of weak references to the object
|
| ----------------------------------------------------------------------
| Class methods inherited from MDAnalysis.analysis.base.AnalysisBase:
|
| get_supported_backends()
| Tuple with backends supported by the core library for a given class.
| User can pass either one of these values as ``backend=...`` to
| :meth:`run()` method, or a custom object that has ``apply`` method
| (see documentation for :meth:`run()`):
|
| - 'serial': no parallelization
| - 'multiprocessing': parallelization using `multiprocessing.Pool`
| - 'dask': parallelization using `dask.delayed.compute()`. Requires
| installation of `mdanalysis[dask]`
|
| If you want to add your own backend to an existing class, pass a
| :class:`backends.BackendBase` subclass (see its documentation to learn
| how to implement it properly), and specify ``unsupported_backend=True``.
|
| Returns
| -------
| tuple
| names of built-in backends that can be used in :meth:`run(backend=...)`
|
|
| .. versionadded:: 2.8.0
|
| ----------------------------------------------------------------------
| Readonly properties inherited from MDAnalysis.analysis.base.AnalysisBase:
|
| parallelizable
| Boolean mark showing that a given class can be parallelizable with
| split-apply-combine procedure. Namely, if we can safely distribute
| :meth:`_single_frame` to multiple workers and then combine them with a
| proper :meth:`_conclude` call. If set to ``False``, no backends except
| for ``serial`` are supported.
|
| .. note:: If you want to check parallelizability of the whole class, without
| explicitly creating an instance of the class, see
| :attr:`_analysis_algorithm_is_parallelizable`. Note that you
| setting it to other value will break things if the algorithm
| behind the analysis is not trivially parallelizable.
|
|
| Returns
| -------
| bool
| if a given ``AnalysisBase`` subclass instance
| is parallelizable with split-apply-combine, or not
|
|
| .. versionadded:: 2.8.0
|
| ----------------------------------------------------------------------
| Methods inherited from maicos.core.base.ProfileBase:
|
| save(self) -> None
| Save results of analysis to file specified by ``output``.
DATA
__all__ = ['DensityCylinder', 'DensityPlanar', 'DensitySphere', 'Diele...
__authors__ = 'MAICoS Developer Team'
VERSION
0.13.dev24+gd60a78220
FILE
/home/runner/work/maicos/maicos/src/maicos/__init__.py
Package-specific page can also be accessed
help(maicos.DensityPlanar)
Help on class DensityPlanar in module maicos.modules.densityplanar:
class DensityPlanar(maicos.core.planar.ProfilePlanarBase)
| DensityPlanar(
| atomgroup: mda.AtomGroup,
| dens: str = 'mass',
| dim: int = 2,
| zmin: float | None = None,
| zmax: float | None = None,
| bin_width: float = 1,
| bin_method: str = 'com',
| grouping: str = 'atoms',
| sym: bool = False,
| refgroup: mda.AtomGroup | None = None,
| unwrap: bool = True,
| pack: bool = True,
| jitter: float = 0.0,
| concfreq: int = 0,
| output: str = 'density.dat'
| ) -> None
|
| Cartesian partial density profiles.
|
| Calculations are carried out for ``mass``
| :math:`(\rm u \cdot Å^{-3})`, ``number`` :math:`(\rm Å^{-3})`, partial ``charge``
| :math:`(\rm e \cdot Å^{-3})` or electron :math:`(\rm e \cdot Å^{-3})` density
| profiles
| along certain cartesian axes ``[x, y, z]`` of the simulation
| cell.
| Cell dimensions are allowed to fluctuate in time.
|
| For grouping with respect to ``molecules``, ``residues`` etc., the corresponding
| centers (i.e., center of mass), taking into account periodic boundary conditions,
| are calculated. For these calculations molecules will be unwrapped/made whole.
| Trajectories containing already whole molecules can be run with ``unwrap=False`` to
| gain a speedup. For grouping with respect to atoms, the ``unwrap`` option is always
| ignored.
|
| For the correlation analysis the central bin
| (:math:`N / 2`) of the 0th's group profile is used. For further information on the correlation analysis please
| refer to :class:`AnalysisBase <maicos.core.base.AnalysisBase>` or the
| :ref:`general-design` section.
|
| Parameters
| ----------
| atomgroup : MDAnalysis.core.groups.AtomGroup
| A :class:`~MDAnalysis.core.groups.AtomGroup` for which the calculations are
| performed.
| dens : {``"mass"``, ``"number"``, ``"charge"``, ``"electron"``}
| density type to be calculated.
|
| dim : {0, 1, 2}
| Dimension for binning (``x=0``, ``y=1``, ``z=1``).
| zmin : float
| Minimal coordinate for evaluation (in Å) with respect to the center of mass of
| the refgroup.
| If ``zmin=None``, all coordinates down to the lower cell boundary are taken into
| account.
| zmax : float
| Maximal coordinate for evaluation (in Å) with respect to the center of mass of the
| refgroup.
| If ``zmax = None``, all coordinates up to the upper cell boundary are taken into
| account.
| bin_width : float
| Width of the bins (in Å).
| bin_method : {``"com"``, ``"cog"``, ``"coc"``}
| Method for the position binning.
|
| The possible options are center of mass (``"com"``), center of geometry (``"cog"``),
| and center of charge (``"coc"``).
| grouping : {``"atoms"``, ``"residues"``, ``"segments"``, ``"molecules"``, ``"fragments"``}
| Atom grouping for the calculations.
|
| The possible grouping options are the atom positions (in the case where
| ``grouping="atoms"``) or the center of mass of the specified grouping unit (in the
| case where ``grouping="residues"``, ``"segments"``, ``"molecules"`` or
| ``"fragments"``).
| sym : bool
| Symmetrize the profile. Only works in combination with ``refgroup``.
| refgroup : MDAnalysis.core.groups.AtomGroup
| Reference :class:`~MDAnalysis.core.groups.AtomGroup` used for the calculation. If
| ``refgroup`` is provided, the calculation is performed relative to the center of
| mass of the AtomGroup. If ``refgroup`` is :obj:`None` the calculations are performed
| with respect to the center of the (changing) box.
| unwrap : bool
| When :obj:`True`, molecules that are broken due to the periodic boundary conditions
| are made whole.
|
| If the input contains molecules that are already whole, speed up the calculation by
| disabling unwrap. To do so, use the flag ``-no-unwrap`` when using MAICoS from the
| command line, or use ``unwrap=False`` when using MAICoS from the Python interpreter.
|
| Note: Molecules containing virtual sites (e.g. TIP4P water models) are not currently
| supported in MDAnalysis. In this case, you need to provide unwrapped trajectory
| files directly, and disable unwrap. Trajectories can be unwrapped, for example,
| using the ``trjconv`` command of GROMACS.
| pack : bool
| When :obj:`True`, molecules are put back into the unit cell. This is required
| because MAICoS only takes into account molecules that are inside the unit cell.
|
| If the input contains molecules that are already packed, speed up the calculation by
| disabling packing with ``pack=False``.
| jitter : float
| Magnitude of the random noise to add to the atomic positions.
|
| A jitter can be used to stabilize the aliasing effects sometimes appearing when
| histogramming data. The jitter value should be about the precision of the
| trajectory. In that case, using jitter will not alter the results of the histogram.
| If ``jitter = 0.0`` (default), the original atomic positions are kept unchanged.
|
| You can estimate the precision of the positions in your trajectory with
| :func:`maicos.lib.util.trajectory_precision`. Note that if the precision is not the
| same for all frames, the smallest precision should be used.
| concfreq : int
| When concfreq (for conclude frequency) is larger than ``0``, the conclude function
| is called and the output files are written every ``concfreq`` frames.
| output : str
| Output filename.
|
| Attributes
| ----------
| results.bin_pos : numpy.ndarray
| Bin positions (in Å) ranging from ``zmin`` to ``zmax``.
| results.profile : numpy.ndarray
| Calculated profile.
| results.dprofile : numpy.ndarray
| Estimated profile's uncertainity.
|
| Notes
| -----
| Partial mass density profiles can be used to calculate the ideal component of the
| chemical potential. For details, take a look at the corresponding :ref:`How-to
| guide<sphx_glr_generated_examples_basics_chemical-potential.py>`.
|
| Method resolution order:
| DensityPlanar
| maicos.core.planar.ProfilePlanarBase
| maicos.core.planar.PlanarBase
| maicos.core.base.AnalysisBase
| maicos.core.base._Runner
| MDAnalysis.analysis.base.AnalysisBase
| maicos.core.base.ProfileBase
| builtins.object
|
| Methods defined here:
|
| __init__(
| self,
| atomgroup: mda.AtomGroup,
| dens: str = 'mass',
| dim: int = 2,
| zmin: float | None = None,
| zmax: float | None = None,
| bin_width: float = 1,
| bin_method: str = 'com',
| grouping: str = 'atoms',
| sym: bool = False,
| refgroup: mda.AtomGroup | None = None,
| unwrap: bool = True,
| pack: bool = True,
| jitter: float = 0.0,
| concfreq: int = 0,
| output: str = 'density.dat'
| ) -> None
| Initialize self. See help(type(self)) for accurate signature.
|
| ----------------------------------------------------------------------
| Readonly properties inherited from maicos.core.planar.PlanarBase:
|
| odims
| Other dimensions perpendicular to dim i.e. (0,2) if dim = 1.
|
| ----------------------------------------------------------------------
| Methods inherited from maicos.core.base.AnalysisBase:
|
| dump(self, filename: str) -> None
| Save analysis state to an ``.npz`` file.
|
| .. warning::
|
| :meth:`dump` is **not** an archival format. The on-disk layout
| tracks the installed MAICoS and MDAnalysis versions and may stop
| loading after an upgrade. Use it to checkpoint or hand off an
| in-progress analysis, not to preserve results long-term. For
| archival output write summary tables with :meth:`save` or export
| :attr:`results` to a stable format.
|
| Persists all statistical accumulators (``means``, ``sems``, ``sums``,
| ``pop``, ``M2``), the ``results`` and ``_obs`` containers, per-frame
| arrays (``timeseries``, ``frames``, ``times``), metadata, the associated
| Universe and the analysed atomgroup. Restore the analysis with :meth:`load`.
|
| Parameters
| ----------
| filename : str
| Path to the output ``.npz`` file.
|
| run(
| self,
| start: int | None = None,
| stop: int | None = None,
| step: int | None = None,
| frames: int | None = None,
| verbose: bool | None = None,
| progressbar_kwargs: dict | None = None
| ) -> Self
| Iterate over the trajectory.
|
| Parameters
| ----------
| start : int
| start frame of analysis
| stop : int
| stop frame of analysis
| step : int
| number of frames to skip between each analysed frame
| frames : array_like
| array of integers or booleans to slice trajectory; ``frames`` can only be
| used *instead* of ``start``, ``stop``, and ``step``. Setting *both*
| ``frames`` and at least one of ``start``, ``stop``, ``step`` to a
| non-default value will raise a :exc:`ValueError`.
| verbose : bool
| Turn on verbosity
| progressbar_kwargs : dict
| ProgressBar keywords with custom parameters regarding progress bar position,
| etc; see :class:`MDAnalysis.lib.log.ProgressBar` for full list.
|
| Returns
| -------
| self : object
| analysis object
|
| savetxt(self, fname: str, X: np.ndarray, columns: list[str] | None = None) -> None
| Save to text.
|
| An extension of the numpy savetxt function. Adds the command line input to the
| header and checks for a doubled defined filesuffix.
|
| Return a header for the text file to save the data to. This method builds a
| generic header that can be used by any MAICoS module. It is called by the save
| method of each module.
|
| The information it collects is:
| - timestamp of the analysis
| - name of the module
| - version of MAICoS that was used
| - command line arguments that were used to run the module
| - module call including the default arguments
| - number of frames that were analyzed
| - atomgroup that was analyzed
| - output messages from modules and base classes (if they exist)
|
| ----------------------------------------------------------------------
| Class methods inherited from maicos.core.base.AnalysisBase:
|
| load(filename: str) -> Self
| Restore an analysis instance from a file created by :meth:`dump`.
|
| Returns a new instance of ``cls`` with all statistical accumulators,
| per-frame arrays, metadata, and the analysed atomgroup populated from
| the file. The rebuilt :class:`MDAnalysis.Universe` carries only the
| topology — no trajectory — so calling :meth:`run` on the returned
| instance raises :class:`RuntimeError`. Use the loaded instance to
| inspect :attr:`results` or to call :meth:`save`.
|
| Parameters
| ----------
| filename : str
| Path to the ``.npz`` file written by :meth:`dump`.
|
| Returns
| -------
| Self
| A new instance of ``cls`` with state restored from ``filename``.
|
| ----------------------------------------------------------------------
| Readonly properties inherited from maicos.core.base.AnalysisBase:
|
| box_center
| Center of the simulation cell.
|
| box_lengths
| Lengths of the simulation cell vectors.
|
| ----------------------------------------------------------------------
| Data descriptors inherited from maicos.core.base._Runner:
|
| __dict__
| dictionary for instance variables
|
| __weakref__
| list of weak references to the object
|
| ----------------------------------------------------------------------
| Class methods inherited from MDAnalysis.analysis.base.AnalysisBase:
|
| get_supported_backends()
| Tuple with backends supported by the core library for a given class.
| User can pass either one of these values as ``backend=...`` to
| :meth:`run()` method, or a custom object that has ``apply`` method
| (see documentation for :meth:`run()`):
|
| - 'serial': no parallelization
| - 'multiprocessing': parallelization using `multiprocessing.Pool`
| - 'dask': parallelization using `dask.delayed.compute()`. Requires
| installation of `mdanalysis[dask]`
|
| If you want to add your own backend to an existing class, pass a
| :class:`backends.BackendBase` subclass (see its documentation to learn
| how to implement it properly), and specify ``unsupported_backend=True``.
|
| Returns
| -------
| tuple
| names of built-in backends that can be used in :meth:`run(backend=...)`
|
|
| .. versionadded:: 2.8.0
|
| ----------------------------------------------------------------------
| Readonly properties inherited from MDAnalysis.analysis.base.AnalysisBase:
|
| parallelizable
| Boolean mark showing that a given class can be parallelizable with
| split-apply-combine procedure. Namely, if we can safely distribute
| :meth:`_single_frame` to multiple workers and then combine them with a
| proper :meth:`_conclude` call. If set to ``False``, no backends except
| for ``serial`` are supported.
|
| .. note:: If you want to check parallelizability of the whole class, without
| explicitly creating an instance of the class, see
| :attr:`_analysis_algorithm_is_parallelizable`. Note that you
| setting it to other value will break things if the algorithm
| behind the analysis is not trivially parallelizable.
|
|
| Returns
| -------
| bool
| if a given ``AnalysisBase`` subclass instance
| is parallelizable with split-apply-combine, or not
|
|
| .. versionadded:: 2.8.0
|
| ----------------------------------------------------------------------
| Methods inherited from maicos.core.base.ProfileBase:
|
| save(self) -> None
| Save results of analysis to file specified by ``output``.
The general help of MAICoS can be accessed using
maicos -h
Package-specific page can also be accessed from the cli
maicos densityplanar -h